

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website https://www.theijcp.org |
Case Report
Volume 13, Number 2, June 2024, pages 54-59

A Five Hundred Six-Gram Giant Orbital Retinoblastoma: An Unusual Case Report
Riccardo Chiorrinia, b, c, Sonia De Francescoa, Mario Fruschellia, Theodora Hadjistilianoua
aDepartment of Medicine, Surgery and Neuroscience, Unit of Ophthalmology, Ocular Oncology, University of Siena, Siena, Italy
bAzienda Ospedaliero-Universitaria Senese, Siena 53100, Italy
cCorresponding Author: Riccardo Chiorrini, Azienda Ospedaliero-Universitaria Senese, Siena 53100, Italy
Manuscript submitted February 20, 2024, accepted March 5, 2024, published online June 23, 2024
Short title: A Case of a Giant Orbital Retinoblastoma
doi: https://doi.org/10.14740/ijcp533
| Abstract | ▴Top |
We present the case of a 14-month-old Caucasian male with advanced orbital retinoblastoma. The patient, having undergone enucleation elsewhere for intraocular retinoblastoma without adjuvant therapy, developed a massive fungating orbital mass shortly after surgery. Despite orbital exenteration and subsequent systemic therapy, the tumor recurred rapidly, leading to systemic metastases and eventual demise. We describe the clinical and diagnostical workup of this rare disease done in a developed country, which given the rarity of this condition in this kind of setting, highlights the importance of education about a prompt approach to avoid the consequences related to a delayed intervention.
Keywords: Retinoblastoma; Orbital retinoblastoma; Orbital tumor; Pediatric orbital tumor
| Introduction | ▴Top |
Retinoblastoma has an incidence of 1:18,000 live births; it accounts for 3% of pediatric tumors and it is the most common intraocular childhood tumor.
Orbital retinoblastoma refers to the extra ocular extension of the lesion that can be detected clinically, radiologically or histopathologically and can be confined to the periocular tissues or can be associated to metastatic spreading.
Primary extraocular extension can occur through various pathways such as direct scleral erosion or involvement of structures like the optic nerve, Schlemm’s canal, posterior ciliary vessels, or nerves, and posterior or anterior emissary channels [1].
Significant risk factors for orbital involvement include delays in diagnosis, late enucleation, and high-risk histopathological features post-enucleation for primary intraocular tumors (including massive choroidal involvement, microscopic extra scleral invasion, and optic nerve involvement beyond the resection margin) [1]. Factors such as prolonged symptom duration (> 6 months), age over 24 months, and secondary glaucoma at presentation increase the likelihood of massive choroidal tumor infiltration [2].
The clinical presentation of the tumor varies based on its size, extent, and the type of orbital retinoblastoma.
Microscopic orbital retinoblastoma often shows no symptoms and is usually diagnosed incidentally during histopathological examination of an enucleated eye removed for intraocular retinoblastoma. Conversely, significant extraocular extension can lead to symptoms resembling a retrobulbar mass [1].
Primary orbital retinoblastoma may present in several ways: with silent proptosis alongside evident intraocular tumor (a common presentation), with rapidly progressive proptosis accompanied by chemosis and inflammation resembling sterile orbital cellulitis, with a palpable episcleral nodule, or in advanced cases, as a prominent fungating mass in the orbit.
Secondary orbital retinoblastoma is suspected when there is extrusion or bulging of the implant or displacement of the ocular prosthesis weeks to years after previous enucleation for intraocular tumor. Orbital recurrence might also manifest as a vascular conjunctival or subconjunctival nodule in the anophthalmic socket, necessitating prosthesis removal for thorough examination and palpation of the socket during follow-up visits [3].
In the current case report, we describe a 14-month-old Caucasian male with intraocular retinoblastoma who was first enucleated in a poor income setting and then, left without the correct treatment, developed a secondary giant orbital mass, so advanced that the tissue hosted worms inside, a peculiar presentation never described in literature. We believe it is important to report this case in order to raise awareness among ophthalmologists in developed countries, where these cases are extremely rare, underscoring the necessity for an immediate diagnosis and a correct management that can save patient’s life.
| Case Report | ▴Top |
A 14-month-old Caucasian male patient presented to our ocular oncology service with an exuberant fungating right orbital mass. The patient was born after a normal conducted pregnancy of 39 weeks, presenting a weight of 3,300 g (-0.15 standard deviation (SD)), height of 51 cm (+0.49 SD), head circumference of 34 cm (+0.42 SD), and Apgar scores were 9, 8 and 9, respectively, at 1, 5 and 10 min. The child was the firstborn of an itinerant family belonging to the Roma community. His parents were not consanguineous and did not have a history of systemic/ocular diseases.
The patient’s right eye had been enucleated in a foreign hospital of the Balkan countries at 9 months of age for advanced intraocular retinoblastoma. The child had received neither post-surgical chemotherapy nor radiotherapy. Soon after enucleation the patient presented to a hospital in Northern Greece because a secondary mass started to grow in the anophthalmic socket, and he was rapidly referred to our clinic. He was not taking any kind of medication at the time he arrived in our clinic.
At first admission, he had a regular pulse of 98 beats per minute (bpm), blood pressure was 107/64 mm Hg, and body temperature was 36.8 °C. He was in a state of dehydration with skin folds. His lips and tongue presented fine fissures. He appeared in a semi-comatose state, and he seldom spoke. His walking ability was altered by the mass weight. In fact, by the time the patient came to our attention, the lesion had reached a size comparable to the child’s head (Fig. 1). Blood tests performed at the presentation showed anemia, with hemoglobin level of 10.4 g/dL, leukocytosis of 15,600/mm3 and eosinophilia of 900/mm3. Other hematocrit values were within normal limits. Hemoconcentration was evidenced by moderate hypernatremia (sodium: 154 mmol/L), hyperkalemia (potassium: 5.8 mmol/L) and hyperosmolality (320 mOsm/kg). Liver and renal indices showed no alterations. Nothing significant emerged from the blood culture. Urinalysis was conducted on a dark yellow urine sample with a strong odor. Urine specific gravity (USG) was 1,033, and osmolality (UOsm) of 770 mOsmol/kg H2O, thus confirmed the concentrated specimen. The pH was 5.6, and the other urinary values were within the normality. Microscopic analysis of urine showed the presence of transitional epithelial cells which had no pathological significance.
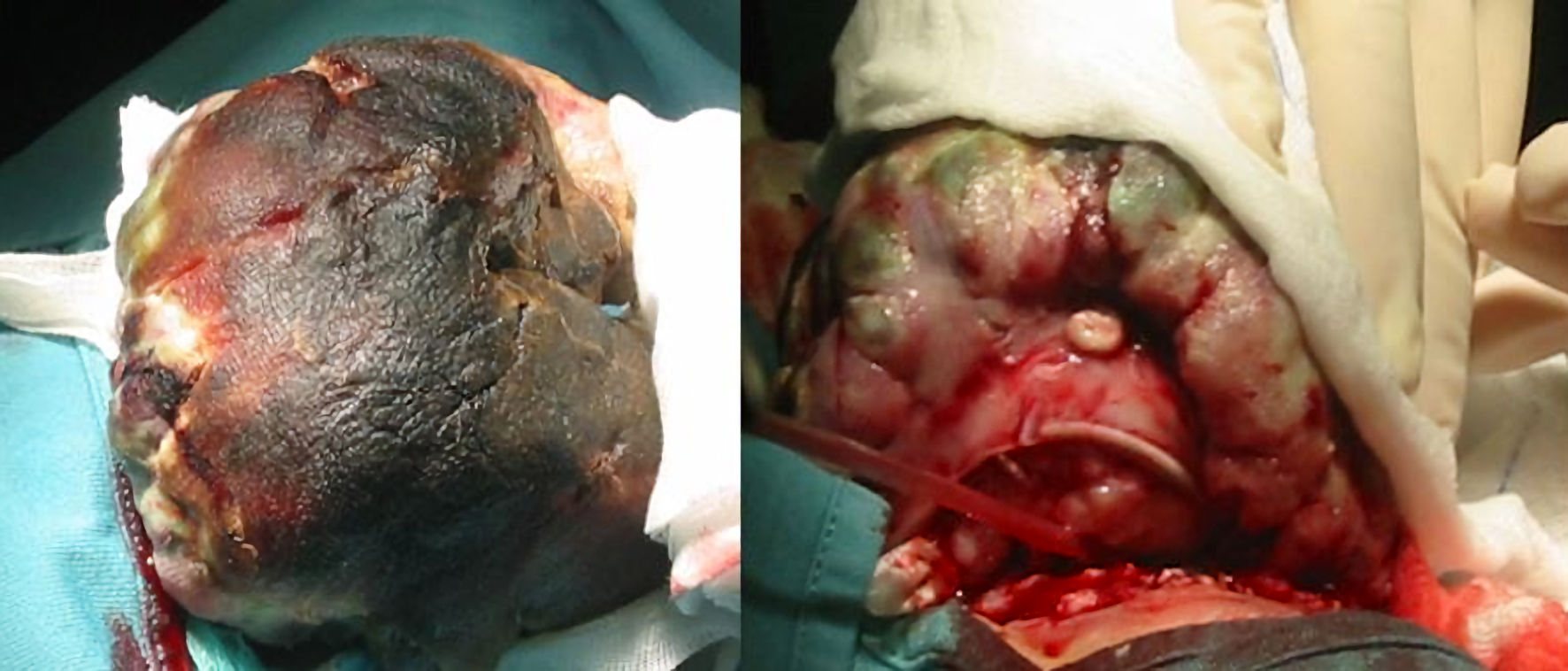 Click for large image | Figure 1. Preoperative pictures. The mass size is comparable to a child’s head. |
The patient underwent mass removal and orbital exenteration. Surgery was performed under general anesthesia and consisted in complete removal of the mass and the entire content of the orbit including both inferior and superior eyelids. It was possible to appreciate a lesion of considerable dimensions (Fig. 2a, b), whose weight was 506 g and dimensions were 12 × 9 × 5 cm. The mass had encephaloid color and consistence and contained suppurative and necrotic tissue, surprisingly hosting worms (Fig. 2c). Histopathological examination showed an undifferentiated retinoblastoma (Fig. 2d), massively infiltrating extrinsic muscles, lacrimal gland, skin and optic nerve stump. The child underwent head and orbit magnetic resonance imaging (MRI) after surgery. As shown in Figure 3a, there was no evidence of tumor residues in the anophthalmic cavity nor invasion in the adjacent structures. Bone scintigraphy, lumbar puncture and bone marrow aspiration were performed, which resulted negative. After orbital exenteration, systemic chemotherapy and radiotherapy were started following the ICE protocol, which involves the combination of ifosfamide, carboplatin and etoposide. Ifosfamide was given at a dose of 2 g/m2 in 4-h infusion on days 1, 2, and 3 (for a total dose of 6 g/m2), associated with Uromitexan and hyperhydration. Uromitexan 700 mg/m2 was given intravenously for 30 min before ifosfamide, and at 2,000 mg/m2 in the following 24 h, together with hyperhydration with 5% glucose solution of 2,000 mL/m2 in 24 h. Etoposide was administered at a dose of 100 mg/m2, diluted in 5% glucose solution to the maximum concentration of 0.4 mg/mL. The divided dose was administered intravenously over 1 h on days 1, 2, and 3. Carboplatin at a dose of 600 mg/m2, diluted in 200 mL/m2 of 5% glucose solution and infused intravenously slowly over 2 h is administered on day 3.
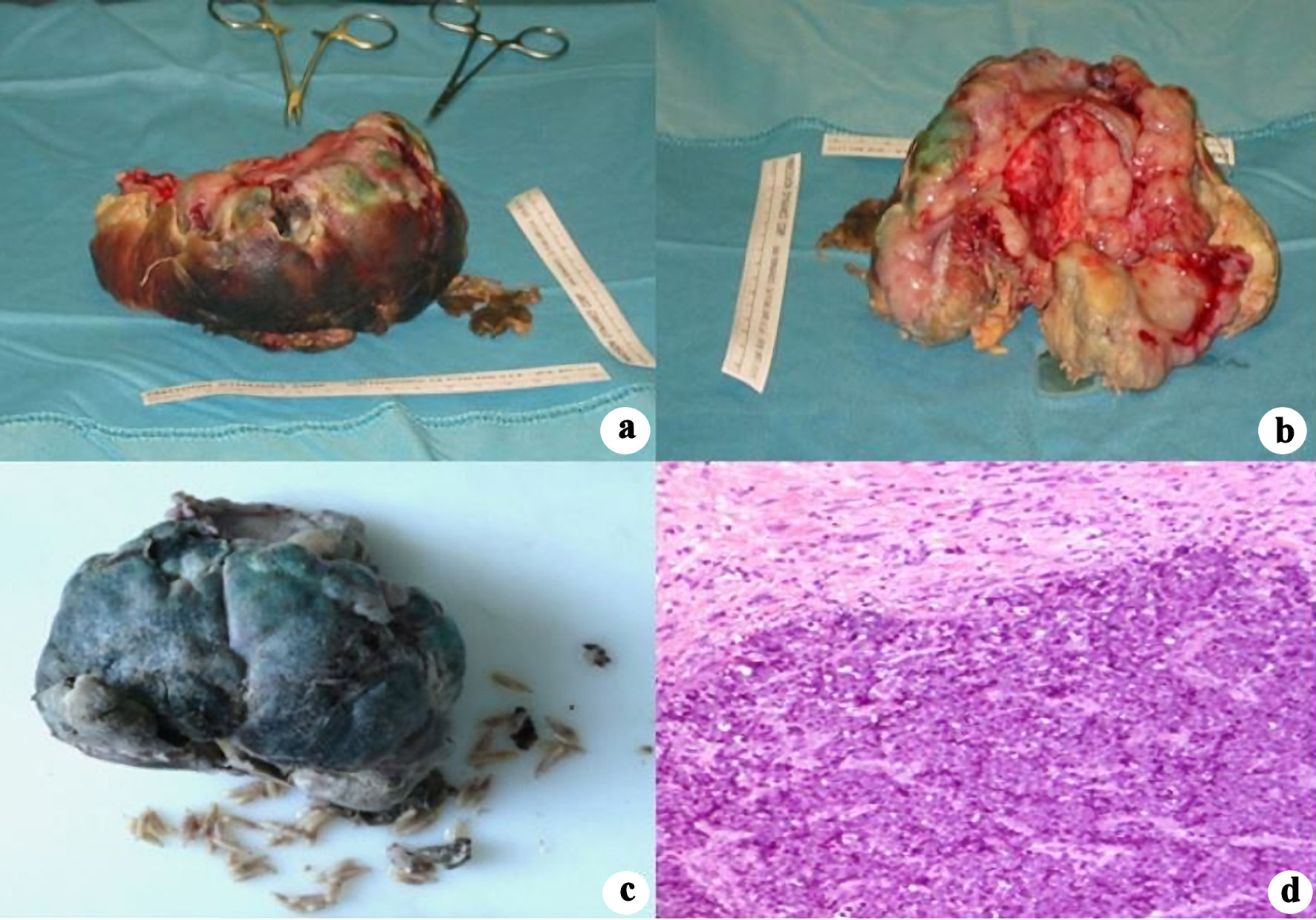 Click for large image | Figure 2. Postoperative pictures demonstrating mass size and its macroscopic features (a, b). Intralesional necrosis and worms (c). Histopathological section showing undifferentiated retinoblastoma (d). |
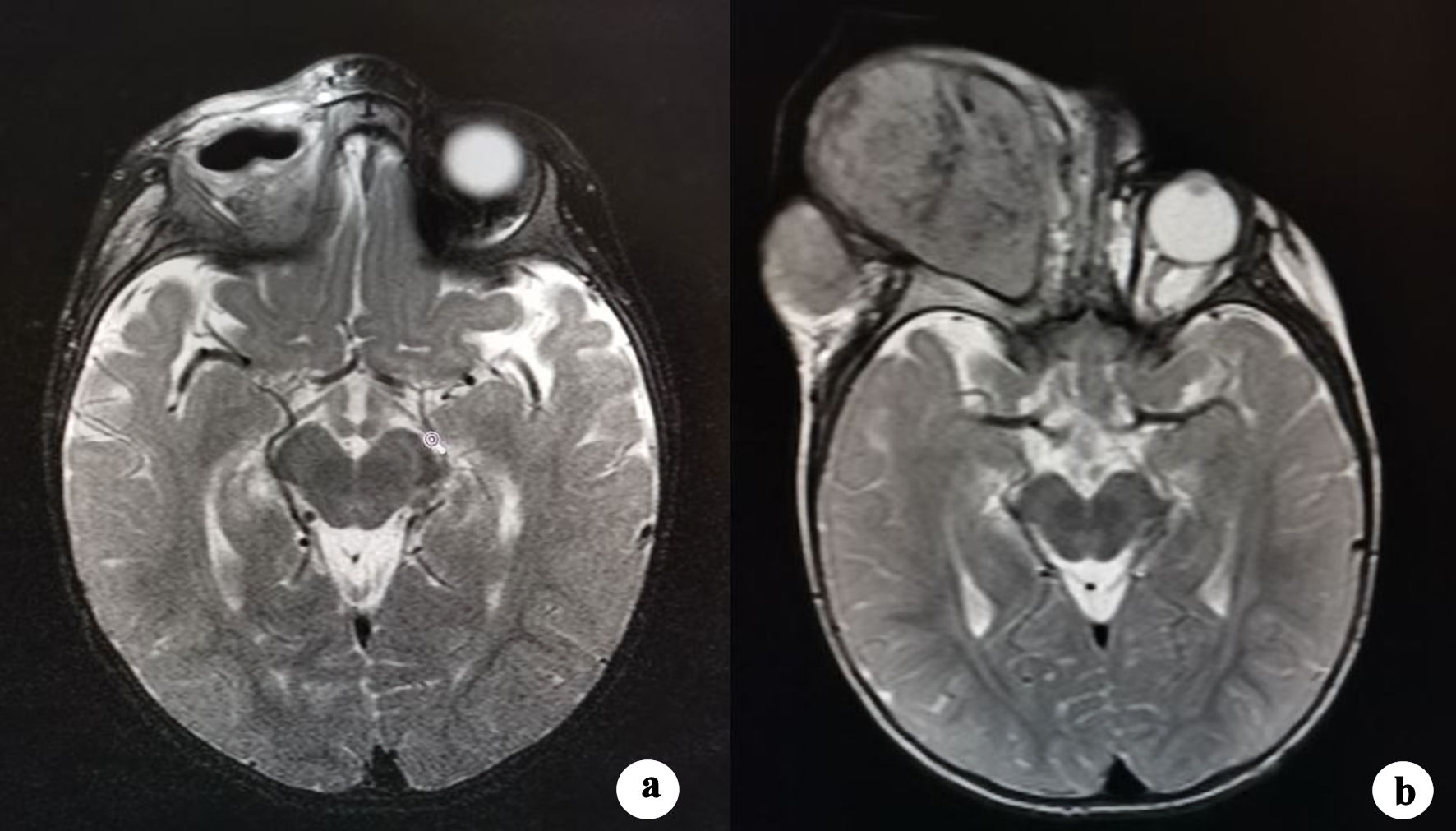 Click for large image | Figure 3. (a) Postoperative T2-weighted MRI scan shows right orbital cavity free from lesions with intact adjacent soft tissues and bones. (b) T2-weighted MRI scan obtained 1 month after the start of the therapy. An amount of effusive isointense tissue has developed in the right orbital cavity, causing its deformation. MRI: magnetic resonance imaging. |
The conditions for starting a chemotherapy cycle were a neutrophil count > 1,000/mm3 and a platelet count > 100,000/mm3, both of which were met in our case.
The therapeutic scheme involves alternating two cycles of ICE with 1 month of local radiotherapy at 4,000 rad, divided into 20 sessions (total time of 1 month), and then repeating two additional cycles of ICE.
Unfortunately, 2 months after the start of therapy, the disease recurred in the orbital cavity. As documented by MRI (Fig. 3b), there was a complete filling of the orbital cavity by the tumor recurrence, with external bulging and extensive infiltration of the surrounding bone structures and soft tissues, with particular interest of the preauricular region.
During discussion of the case within the multidisciplinary oncology group with the child’s parents, the family decided to interrupt therapy and returned to their country of origin, being aware of the poor prognosis. We have been informed that the child died 3 months after for the presence of multiple brain metastases.
| Discussion | ▴Top |
This report describes a 14-months-old child affected by a secondary orbital retinoblastoma who was first enucleated in a low-income setting at the age of 1 month. The patient has undergone mass removal and orbital exenteration in our clinic. The detection, in the context of a high-income setting, of a 506-g fungating mass with worms inside makes our case unique. As we know so far, this has not been described in literature. Despite our clinical experience decades in the field of retinoblastoma, the management of the case did not lead to the survival of the patient. This highlights the importance of early diagnosis and correct classification if similar cases arise in more developed contexts, in which such lesions are extremely rare, and failure to identify them can have a critical impact on the prognosis.
In fact, orbital extension is uncommon in developed nations, occurring in approximately 6.3% to 7.6% of cases [4, 5], typically manifesting as a recurrence following previous enucleation for primary intraocular tumors (known as secondary orbital retinoblastoma) [6]. However, in less developed regions, where access to medical care is limited and cultural beliefs may resist enucleation, primary orbital retinoblastoma is a significant presentation, comprising up to 18% of newly diagnosed cases in Mexico, 36% in Taiwan, and 40% in Nepal [7-9]. The lack of education and impossibility to access adequate medical care causes diagnostic delay, late enucleation, extraocular and systemic spread and subsequent poor prognosis [10]. In the differential diagnosis of orbital invasion, we can consider eyelid malignancies (e.g., basal cell carcinoma, squamous cell carcinoma, sebaceous gland carcinoma) or conjunctival tumors (e.g., melanoma). All the mentioned lesions are typically tumors of the adult, so the age of presentation and/or the previous eye history easily guide the diagnostic process as in our report.
According to the eighth edition of the American Joint Committee on Cancer (AJCC) staging for retinoblastoma, extraocular retinoblastoma belongs to cT4 stage. CT4a indicates radiologic evidence of retrobulbar optic nerve involvement or thickening of optic nerve or involvement of orbital tissues, while cT4b signifies extraocular tumor, clinically evident with proptosis and/or an orbital mass, as in our case. CM1 investigates clinical signs of distant metastasis. Grade cM1a is assigned to tumors involving any distant site (e.g., bone marrow, liver) on clinical or radiologic tests; grade cM1b is for tumors involving the central nervous system on radiologic imaging [11]. This system is important because it can be used to predict metastases-related mortality with a survival rate of 45% at 5 years in T4 stage disease [12].
Patients with orbital retinoblastoma extension require diagnostic investigations which are mandatory to evaluate the extent of the tumor, providing a correct classification and the consequent risk of metastases. These include evaluation under anesthesia, computed tomography (CT) and MRI of the head and orbit. CT scans typically reveal an intraconal mass with moderate contrast enhancement. On T1-weighted MRI images, orbital retinoblastoma appears hyperintense compared to the vitreous and muscles, and hypointense compared to fat, while T2-weighted images show a mass that is hypointense relative to the vitreous and isointense relative to fat. Additionally, a metastasis workup is essential, involving palpation of regional lymph nodes and subsequent fine needle aspiration biopsy (FNAB) if involvement is suspected, technetium-99 bone scintigraphy, lumbar puncture with cerebrospinal fluid cytology, bone marrow aspiration, and positron emission tomography coupled with computed tomography (PET-CT) [7, 11].
Treatment protocols for orbital retinoblastoma lack universal consensus but typically involve multimodal therapy with neoadjuvant chemotherapy followed by orbital exenteration, adjuvant chemotherapy, and external beam radiotherapy. There are numerous reasons for this: 1) Sole reliance on systemic chemotherapy is improbable to completely eliminate residual orbital disease; 2) Relying solely on orbital exenteration is unlikely to achieve complete surgical clearance; 3) External beam radiotherapy alone is unlikely to prevent systemic metastasis.
Retinoblastoma is a highly chemosensitive tumor that responds well to many chemotherapeutic agents such as cyclophosphamide, platinum derivatives, adriamycin, vincristine, and epipodophyllotoxins demonstrate high efficacy [6, 13].
Chantada et al [14] published various chemotherapy regimens recommended by SIOP-PODC, tailored to different income levels. For upper-middle income countries, three potential chemotherapy protocols are suggested for preoperative, adjuvant therapy in orbital retinoblastoma, and metastatic disease treatment: 1) Carboplatin (500 mg/m2 on days 1 - 2) + etoposide (100 mg/m2 on days 1 - 3); 2) Cyclophosphamide (65 mg/kg on day 1) + vincristine (1.5 mg/m2 on day 1) + idarubicin (10 mg/m2 on day1); 3) Ifosfamide (1.8 g/m2 on days 1 - 5) + etoposide (100 mg/m2 on days1 - 5) +/- carboplatin (400 mg/m2 on days 1 and 2). For low-income or lower-middle income settings, the recommended regimens are: 1) Carboplatin (500 - 560 mg/m2 on day 1) + etoposide (100 - 150 mg/m2 on days 1 - 2) + vincristine (1.5 mg/m2 on day 1); 2) Cyclophosphamide (40 mg/kg on day 1) + vincristine (1.5 mg/m2 on day 1) + doxorubicin (30 mg/m2 on day 1).
Autologous stem cell transplantation can complement these protocols when high-dose chemotherapy regimens lead to myelotoxicity [15].
Intrathecal chemotherapy with cytarabine or topotecan might be considered in cases of leptomeningeal dissemination, although evidence supporting this is limited [16].
Historically, orbital retinoblastoma prognosis was poor, particularly with macroscopic involvement, correlating with higher systemic metastasis risk and mortality rates [17] (10 - 27 times higher risk of systemic metastasis and a mortality of 90% at 10 years), while microscopic involvement has a better prognosis. However, recent decades have witnessed improved outcomes attributed to advancements in surgical techniques, multidisciplinary approaches, and combination therapies.
Conclusions
The poor prognosis in our patient can be attributed to the delay in presentation and the lack of post-enucleation adjuvant treatment, and therefore underscores several critical points in the management of orbital retinoblastoma which can be considered as take-home messages of our report.
Firstly, a careful interpretation of the histological sample at the time of enucleation is mandatory, given that the basis of such a development was certainly an infiltration of the sclera and/or the optic nerve. For these reasons a high expertise in pathology service and an immediate start of therapy could have saved the child’s life.
Secondly, the rarity and aggressiveness of the disease in a developed country highlight the need for heightened awareness among ophthalmologists regarding its prompt recognition and appropriate therapeutic approach. Although uncommon, cases like this emphasize the importance of considering orbital retinoblastoma in the differential diagnosis of pediatric orbital masses, especially in patients with a history of intraocular retinoblastoma.
Thirdly, the rapid recurrence and metastatic spread observed in this case underscore the urgent need for multidisciplinary management strategies. Despite aggressive surgical intervention and adjuvant therapy, disease control was not achieved, emphasizing the challenges inherent in managing advanced cases of orbital retinoblastoma, in special consideration in countries with poor clinical experience about this rare entity. Clinical figures such as the pathologist and the neuroradiologist are fundamental for correct staging, therapeutic approach, and follow-up of retinoblastoma.
In conclusion, this case emphasizes the complexities in the management of aggressive orbital retinoblastoma. It highlights the importance of timely diagnosis, comprehensive treatment, and multidisciplinary collaboration to optimize outcomes for patients with this devastating malignancy. Furthermore, it underscores the need for continued research and medical education to improve our understanding and management of this rare but potentially life-threatening condition.
Acknowledgments
The authors wish to thank Doctor Paola Gennari and Doctor Matteo Zanoni (Unit of Neuroradiology, University of Siena) for their advice and support.
Financial Disclosure
This study received no specific grant from any funding agency in the public, commercial, or not-for profit sectors.
Conflict of Interest
The authors declare that there is no conflict of interest with the themes reported in this clinical case.
Informed Consent
The authors did not deem it necessary to obtain written informed consent given the impossibility of tracing the patient, considering the information provided and the absence of facial images.
Author Contributions
Professor Hadjistilianou and Dr. Chiorrini wrote the first draft of the manuscript. Professor Hadjistilianou, Dr. Chiorrini, Dr. De Francesco and Professor Fruschelli reviewed the manuscript and obtained all the ophthalmological images reported in the case.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Nema HV, Nema N. Ocular tumors. Ocul Tumors. 2020;1-260.
- Kaliki S, Tahiliani P, Iram S, Ali MH, Mishra DK, Reddy VA. Choroidal infiltration by retinoblastoma: predictive clinical features and outcome. J Pediatr Ophthalmol Strabismus. 2016;53(6):349-356.
doi pubmed - Balasopoulou A, Κokkinos P, Pagoulatos D, Plotas P, Makri OE, Georgakopoulos CD, et al. Symposium recent advances and challenges in the management of retinoblastoma globe - saving treatments. BMC Ophthalmol [Internet]. 2017;17(1):1.
doi pubmed pmc - Grabowski EF, Abramson DH. Intraocular and extraocular retinoblastoma. Hematol Oncol Clin North Am. 1987;1(4):721-735.
pubmed - Kumari N, Das S. Orbital retinoblastoma. Retin Diagnosis to Manag. 2022;LXXII:263-279.
- Doz F, Khelfaoui F, Mosseri V, Validire P, Quintana E, Michon J, Desjardins L, et al. The role of chemotherapy in orbital involvement of retinoblastoma. The experience of a single institution with 33 patients. Cancer. 1994;74(2):722-732.
doi pubmed - Leal-Leal C, Flores-Rojo M, Medina-Sanson A, Cerecedo-Diaz F, Sanchez-Felix S, Gonzalez-Ramella O, Perez-Perez F, et al. A multicentre report from the Mexican Retinoblastoma Group. Br J Ophthalmol. 2004;88(8):1074-1077.
doi pubmed pmc - Chen PY, Kao LY, Chao AN, Wu WC, Sun MH, Su WW, Liu CH. Retinoblastoma in Taiwan: survival and clinical characteristics. Jpn J Ophthalmol. 2021;65(4):546-553.
doi pubmed - Badhu B, Sah SP, Thakur SK, Dulal S, Kumar S, Sood A, Das H, et al. Clinical presentation of retinoblastoma in Eastern Nepal. Clin Exp Ophthalmol. 2005;33(4):386-389.
doi pubmed - Honavar SG, Manjandavida FP, Reddy VAP. Orbital retinoblastoma: an update. Indian J Ophthalmol. 2017;65(6):435-442.
doi pubmed pmc - TNM8: The updated TNM classification for retinoblastoma. Community Eye Health. 2018;31(101):34.
pubmed pmc - Tomar AS, Finger PT, Gallie B, Mallipatna A, Kivela TT, Zhang C, Zhao J, et al. A multicenter, international collaborative study for american joint committee on cancer staging of retinoblastoma: part I: metastasis-associated mortality. Ophthalmology. 2020;127(12):1719-1732.
doi pubmed - Amendola BE, Lamm FR, Markoe AM, Karlsson UL, Shields J, Shields CL, Augsburger J, et al. Radiotherapy of retinoblastoma. A review of 63 children treated with different irradiation techniques. Cancer. 1990;66(1):21-26.
doi pubmed - Chantada G, Luna-Fineman S, Sitorus RS, Kruger M, Israels T, Leal-Leal C, Bakhshi S, et al. SIOP-PODC recommendations for graduated-intensity treatment of retinoblastoma in developing countries. Pediatr Blood Cancer. 2013;60(5):719-727.
doi pubmed - Palma J, Sasso DF, Dufort G, Koop K, Sampor C, Diez B, Richard L, et al. Successful treatment of metastatic retinoblastoma with high-dose chemotherapy and autologous stem cell rescue in South America. Bone Marrow Transplant. 2012;47(4):522-527.
doi pubmed - Blaney SM, Heideman R, Berg S, Adamson P, Gillespie A, Geyer JR, Packer R, et al. Phase I clinical trial of intrathecal topotecan in patients with neoplastic meningitis. J Clin Oncol. 2003;21(1):143-147.
doi pubmed - Singh AD, Shields CL, Shields JA. Prognostic factors in retinoblastoma. J Pediatr Ophthalmol Strabismus. 2000;37(3):134-141; quiz 168-139.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.