

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website https://www.theijcp.org |
Case Report
Volume 9, Number 4, December 2020, pages 130-134
Insights Into Management of Camurati-Engelmann Disease: A Case Series of Three Siblings
W. Hunter Slempa, Janel D. Hunterb, Elizabeth T. Walshb, Cathrine Constantacosb, David F. Crudob, c
aSection of General Pediatrics, Department of Pediatrics, Wake Forest University School of Medicine, Winston-Salem, NC, USA
bSection of Pediatric Endocrinology, Department of Pediatrics, Wake Forest University School of Medicine, Winston-Salem, NC, USA
cCorresponding Author: David F. Crudo, Department of Pediatrics, Wake Forest University School of Medicine, 1 Medical Center Boulevard, Winston-Salem, NC 27157, USA
Manuscript submitted August 24, 2020, accepted September 12, 2020, published online November 3, 2020
Short title: Management of Camurati-Engelmann Disease
doi: https://doi.org/10.14740/ijcp403
| Abstract | ▴Top |
Camurati-Engelmann disease (CED) is an autosomal dominant skeletal dysplasia characterized by progressive sclerosis of long bones due to a mutation in the transforming growth factor beta-1 gene. Patients experience progressive pain, weakness, and fatigability over time. There are no consensus guidelines for treatment though the use of several types of glucocorticoids, angiotensin II receptor blockers, and other therapies have been described. We present the cases of three siblings with CED managed with different treatment modalities over time (prednisone, losartan, and deflazacort). We provide objective data (pain scores, walk-test results, erythrocyte sedimentation rates) to demonstrate therapeutic efficacy. Prednisone resulted in the greatest improvement in pain; however, its use was limited by significant weight gain. Deflazacort was successful in improving pain and fatigability without the weight gain. Risks and benefits must be considered carefully as the cost of deflazacort is significantly higher than prednisone or losartan.
Keywords: Camurati-Engelmann; Glucocorticoid; Losartan; Deflazacort
| Introduction | ▴Top |
Camurati-Engelmann disease (CED) is a rare form of skeletal dysplasia characterized by progressive sclerosis of long bones that was initially described in a trio of papers in the 1920s [1-3]. CED has an autosomal dominant inheritance pattern and arises from missense mutations in the transforming growth factor beta-1 (TGFB1) gene that produces transforming growth factor beta-1 (TGFβ-1) protein. Mutations typically cause a gain-of-function in TGFβ-1 that disrupts the regulation of bone turnover. The increase in TGFβ-1 activity is postulated to stimulate bone formation while inhibiting resorption, leading to the characteristic radiographic findings of progressive hyperostosis and cortical thickening of the long bones and skull, predominately involving the diaphysis with epiphyseal sparing [4, 5]. These changes manifest clinically with extremity pain, muscular weakness, diminished exercise reserve and decreased fat stores. Patients often have a characteristic waddling gait and may experience various cranial nerve palsies due to hyperostosis of the skull [4-6].
There are no consensus guidelines for optimal treatment of CED; however, glucocorticoids and losartan are the most utilized modalities reported in the literature. Bisphosphonates and nonsteroidal anti-inflammatory drugs have also been used with limited efficacy [7-9]. These therapies have demonstrated varying levels of success in pediatric patients and data are scarce given the rarity of this condition.
We present three siblings with CED with similar phenotypic disease. Each patient experienced varying levels of symptomatic relief and improvement in functional status with prednisone, losartan, or deflazacort, with the greatest improvement using prednisone. Unfortunately, a side effect of excessive weight gain limited the duration of prednisone therapy. Symptomatic relief was achieved to a somewhat lesser extent with losartan or deflazacort without the untoward side effect of weight gain. These cases provide further insight into pharmacological management of CED.
| Case Reports | ▴Top |
Case 1
Case 1 is a 17-year-old Syrian man from non-consanguineous parents, who presented with lower extremity pain, proximal muscle weakness, and muscular wasting. His symptoms were attributed initially to a muscular dystrophy of unknown etiology. Physical exam was notable for a very thin man with short stature (World Health Organization (WHO) -2.27 standard deviation score (SDS) height, -4.15 SDS Body Mass Index (BMI)), decreased muscle mass in upper and lower extremities, lack of subcutaneous fat, and pain to palpation of all extremities. Lower extremity radiographs demonstrated marked osteopenia and patchy lucencies scattered along the tibia and fibula bilaterally, as well as medial bowing of the fibula. Osseous survey revealed generalized osteopenia that was most pronounced in long bones, with sparing of skull (see Figure 1 for radiographs). Spine radiographs showed 14 degrees of thoracolumbar dextroscoliosis. Dual-energy X-ray absorptiometry confirmed osteopenia with bone mineral density z-scores of -3.6 and -3.5 of the lumbar spine and total body less head respectively. He initially received 2,000 IU cholecalciferol and 1,200 mg elemental calcium daily to treat osteopenia and vitamin D insufficiency. His pain and weakness worsened over time, necessitating a period of wheelchair use. Due to the worsening symptoms and a new concern for occult lower extremity fracture, his osteopenia was treated with two bisphosphonate infusions (zoledronic acid, 0.0125 mg/kg/dose) 7 weeks apart. He did experience short-lived periods of reduced pain following the infusions. Genetics consultation was obtained and the diagnosis of CED was made based on clinical symptoms, radiographic findings, and family history. The diagnosis of CED was confirmed with genetic testing which revealed a heterozygous mutation in TGFB1 (c.652C>T, p.Arg218Cys). His father was subsequently shown to have the same mutation.
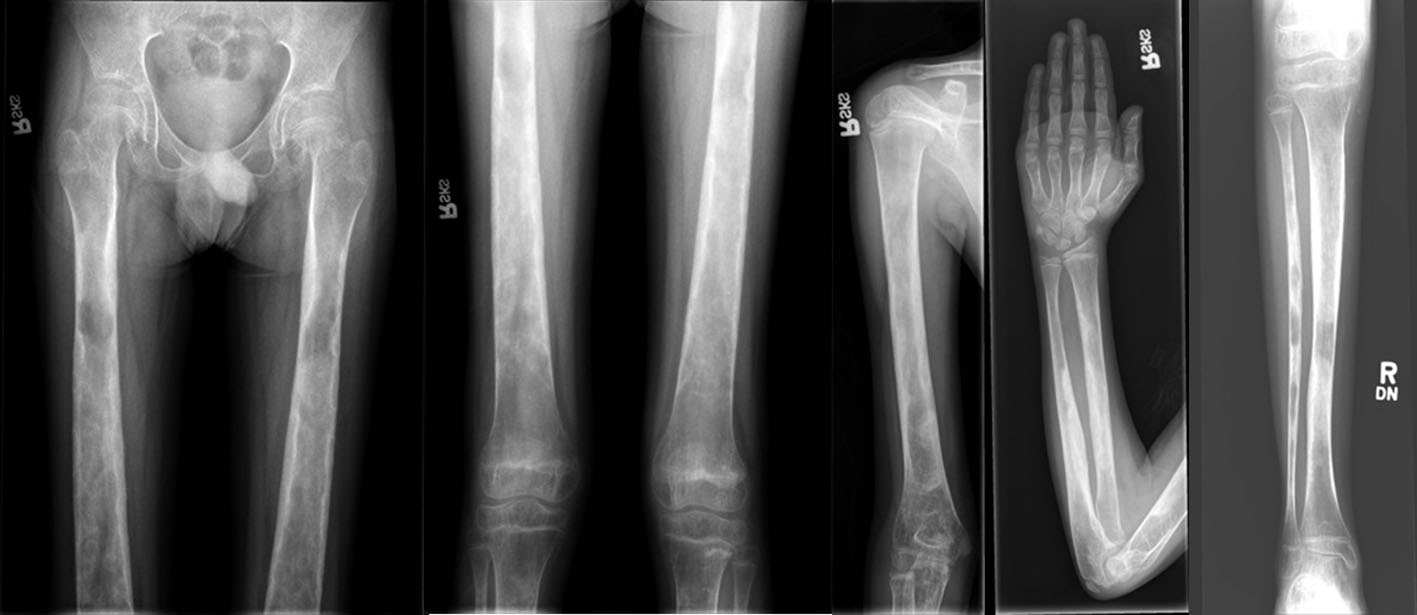 Click for large image | Figure 1. Lower extremities/upper extremities: generalized osteopenia with cortical thickening and foci of sclerosis within the diaphyses of the long bones. |
He was started on a once daily dose of prednisone 30 mg (0.95 mg/kg), and his pain was monitored using the analog Wong-Baker FACES® scale (0 is no pain, 10 is the worst possible pain). He reported 4 of 10 pain at the initiation of prednisone which decreased to 2 of 10 and ultimately resolved after 3 months of therapy. Erythrocyte sedimentation rate (ESR) was initially elevated to 56 mm/h and normalized to 16 mm/h on therapy. Unfortunately, he experienced significant weight gain of 8.4 kg (+ 26% from baseline) over 3 months of prednisone use and was subsequently transitioned to once daily losartan 37.5 mg (0.94 mg/kg). As the prednisone dose was weaned, he experienced recurrence of leg pain, which he rated a 4 of 10. The same level of pain persisted on losartan therapy and worsened to 8 of 10 during a brief period of discontinuation of losartan therapy due to a medication safety recall. He was then transitioned to once daily deflazacort 30 mg (0.70 mg/kg), a newer glucocorticoid with a favorable side effect profile over prednisone. Treatment with deflazacort improved pain to a score of 2 of 10 and he reported subjective improvement in his ability to ambulate. His pain increased over time but responded well to an increase in deflazacort dosing to 39 mg (1.13 mg/kg). He has not experienced weight gain while on deflazacort. Table 1 summarizes his clinical course.
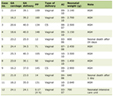 Click to view | Table 1. Clinical Course on Therapy |
Case 2
Case 2 is the 18-year-old male sibling of case 1 who was referred to Pediatric Endocrinology clinic for evaluation of metabolic bone disease and vitamin D deficiency. Symptoms were initially attributed to the same unidentified type of muscular dystrophy that affected his brother and father. His clinical presentation was notable for pain throughout his hips, thighs, and knees that was most significant in the morning. He also reported weakness and “tiring easily”. Physical exam was notable for a very thin man with short stature (WHO -2.82 SDS height, -6.67 SDS BMI) and diminished musculature in the upper and lower extremities. He had little subcutaneous fat. He demonstrated a wide-based, waddling gait. He had decreased strength in the upper and lower extremities and joint contractures. He had no history of fractures or early tooth loss, although he did have significant dental decay and a broken tooth at the time of his presentation. He also had a history of recurrent nephrolithiasis. A presumed diagnosis of CED was made following the positive genetic testing in his sibling with a similar phenotype and characteristic long bone radiographic appearance. He was subsequently found to have the same heterozygous mutation in TGFB1 (c.652C>T, p.Arg218Cys) as his brother and father. At his presentation he was taking vitamin D 50,000 IU weekly for a recent diagnosis of vitamin D deficiency (vitamin D 8 ng/mL). Using the Wong-Baker FACES® pain scale, he rated his hip, thigh, and knee pain as 10 of 10 in the morning with an improvement to 8 of 10 later in the day. He was able to walk 354 m in 6 min. Initial labs were notable for an elevated ESR of 74 mm/h. He was started on prednisone 25 mg (1.08 mg/kg) once daily and reported improvement in pain within days to a score of 2 of 10 with increased ability to ambulate. Three months into therapy his pain had fully resolved and he was able to resume playing soccer. His ESR had also normalized. He also experienced a significant weight gain of 4.2 kg (+ 18% from baseline) over 3 months of therapy. His prednisone was decreased to 15 mg (0.55 mg/kg) and losartan was initiated at a once daily dose of 25 mg (0.91 mg/kg). He continued to have good pain control on this regimen but experienced additional weight gain, so he was weaned from prednisone. Discontinuation of prednisone resulted in return of leg pain so deflazacort was recommended. Unfortunately, a losartan recall and difficulty in obtaining authorization for deflazacort resulted in a gap in therapy, during which he experienced worsening pain. Initiation of once daily deflazacort 18 mg (0.65 mg/kg)resulted in a modest improvement in pain. A repeat 6-min walk test showed a 15% increase in distance traveled. Table 1 summarizes his clinical course.
Case 3
Case 3 is the 7-year-old female sibling of cases 1 and 2. She presented to Pediatric Endocrinology clinic with the presumed diagnosis CED based on symptoms and family history. Prior to that presentation her symptoms of leg muscle weakness and pain were attributed to the same unidentified type of muscular dystrophy that affected her brothers and father. Her phenotype was similar to that of her siblings with characteristic long bone changes on radiographs, osteopenia and poor linear growth. She was subsequently found to have the same heterozygous mutation in TGFB1 (c.652C>T, p.Arg218Cys) as her brothers and father. She had no history of fractures. Her exam was significant for a thin woman with short stature (WHO -1.70 SDS height, -2.97 SDS BMI), as well as decreased muscle mass and strength. At her presentations she was currently taking vitamin D 1,000 IU daily and acetaminophen as needed for leg pain. Using the Wong-Baker FACES® pain scale, she initially reported 6 of 10 extremity pain. Laboratory evaluation demonstrated vitamin D deficiency and elevated ESR of 42 mm/h. She traveled 422 m on her initial 6-min walk test. She was started on prednisone 15 mg (1.11 mg/kg) once daily and her pain and ability to walk both improved. Her ESR normalized to 11 mm/h. She also experienced a significant weight gain of 2.5 kg (+ 18% from baseline) over 3 months of prednisone therapy, prompting a 50% decrease in her prednisone dose and the addition of losartan 12.5 mg (0.78 mg/kg). She had continued good control of her pain on this regimen. Prednisone taper resulted in slowing of her weight gain, but she experienced increasing pain while on losartan monotherapy. During the transient discontinuation of losartan therapy due to a medication safety recall, she experienced an increase in pain, which she rated a 6 of 10. She was then started on once daily deflazacort 9 mg (0.66 mg/kg) with improvement in her pain score to 2 of 10 after 1 month of therapy. Her distance travelled in a 6-min walk test improved by 16%. Table 1 summarizes her clinical course.
| Discussion | ▴Top |
CED is a rare progressive skeletal dysplasia with variable presentation even within families who share a common genetic mutation. Clinical presentation may range from patients with debilitating pain and weakness with increased intracranial pressure and paresis from hyperostosis of the skull to asymptomatic adults diagnosed incidentally via radiographs [6]. While there are no standard guidelines for therapy of CED, the use of nonsteroidal anti-inflammatory drugs, acetaminophen, bisphosphonates, glucocorticoids and losartan has been described [7-10]. There is also a report of improvement in pain in a patient with CED undergoing anti-tumor necrosis factor alpha therapy for uncreative colitis [11]. The current literature favors glucocorticoids and losartan as the most successful therapies to date [10].
Glucocorticoids have been shown to reduce pain and markers of inflammation most consistently in patients with CED [10]; however, the rationale for their efficacy remains unknown. Glucocorticoids are known to inhibit osteoblast proliferation, but histological studies following steroid therapy have also demonstrated increased bone resorption and osteoblast activity due to secondary remodeling. Glucocorticoids may also inhibit factors responsible for TGFβ-1 stimulated muscle fibrosis [12]. Prednisone has typically been used in many cases, but its untoward side effects (growth impairment in children, worsening osteopenia, and weight gain) may limit its duration of therapy. Newer glucocorticoid formulations like deflazacort are now being used more widely. Deflazacort, a derivative of prednisone with a more favorable side-effect profile, has been used successfully in other disorders requiring chronic steroid therapy such as muscular dystrophy. Deflazacort is significantly costlier than prednisone, so risks and benefits must be weighed.
Losartan, an angiotensin II receptor blocker, downregulates the expression of TGFβ-1 receptors, and has been employed in the treatment of CED due to its anti-TGFβ-1 properties [8]. There are existing reports which show not only resolution of pain but also improvement in bone mineral density and fat mass accrual [8, 13, 14]. Other reports show no improvement with losartan [12].
Bisphosphonate therapy has been attempted due to the presence of increased markers of bone turnover (osteocalcin, procollagen type 1 N-terminal propeptide, and cross-linked carboxyterminal telopeptide of type 1 collagen) found in patients with CED. Zoledronic acid has been shown to normalize these markers of bone turnover and result in pain improvement in some patients [15]. Nevertheless, its use is disputed as there are reports of worsening pain in other patients [12].
In our cases, prednisone resulted in the most profound improvement in symptoms, leading to complete resolution of extremity pain in two of three cases within 3 months of initiation of therapy. In the third patient, there was complete resolution of pain with the addition of losartan to prednisone therapy after 4 months. All three patients have demonstrated a reduction in ESR as a marker of inflammation and an improvement in functional status as measured by a 6-min walk test. Improvement in ambulation was most notable in case 1, where in response to initiation of prednisone the patient regained the ability to ambulate independently after requiring the use of a wheelchair. Other published cases also report complete resolution of pain after 1 - 3 months of deflazacort therapy [7]. While prednisone was most successful in ameliorating symptoms, side effects of significant weight gain limited the duration of its use. Fortunately, our patients did not experience hypertension or Cushingoid features.
Losartan therapy was used alone and in combination with corticosteroids in our cases. Unfortunately, our patients experienced recurrence of pain on losartan monotherapy. Losartan was discontinued ultimately in all of our patients due to an untimely safety recall of that medication.
All of our patients experienced improved pain with the initiation of deflazacort, as well as reduction in ESR. It is worth noting that pain reduction was slightly less than with prednisone. Nevertheless, quality of life and functional status were improved and they did not experience excessive weight gain.
Conclusions
We present this series of three related patients with variable responses to three therapeutic interventions in order to provide objective insight into the pharmacological management of CED. Though our patients’ clinical presentations were similar and their genotypes were identical, their responses to the various treatment modalities slightly differed. It is possible that response to therapy may relate in some way to the variable penetrance of symptoms demonstrated within families with CED, although this remains unknown [6].
There are no consensus guidelines for the management of CED with glucocorticoids and losartan having been shown to be effective in many cases [4-10, 14], although patient response is variable. Our experience supports dramatic improvement in pain and mobility with glucocorticoid therapy, to a slightly lesser extent with losartan, and provides objective data supporting this clinical improvement.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
The patients and/or their families were informed that data from the case would be submitted for publication and gave their consent.
Author Contributions
DFC was the primary physician providing care for these patients, conceptualized the manuscript, and is the guarantor. JDH, ETW, and CC also provided care to the patients. WHS, JDH and DFC collaborated in writing the manuscript. All authors revised the paper critically for intellectual content and approved the final version. All authors agree to be accountable for the work and to ensure that any questions relating to the accuracy and integrity of the paper are investigated and properly resolved.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Cockayne EA. Case for diagnosis. Proc R Soc Med. 1920;13(Sect Study Dis Child):132-136.
doi - Camurati M. Di un raro caso di osteite simmetrica ereditaria degli arti inferiori. Chir Organi Mov. 1922;6:662-665 (in Italian).
- Engelmann G. Ein fall von ostheopathia hyperostotica (sclerotisans) multiplex infantilis. Forschritte auf dem Gebiete der Rontgenstrahlen und der Nuklearmedizin. 1929;39:1101-1106 (in German).
- Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, Ralston S, Guanabens N, et al. Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet. 2006;43(1):1-11.
doi pubmed - Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-beta1 to the bone. Endocr Rev. 2005;26(6):743-774.
doi pubmed - Wallace SE, Lachman RS, Mekikian PB, Bui KK, Wilcox WR. Marked phenotypic variability in progressive diaphyseal dysplasia (Camurati-Engelmann disease): report of a four-generation pedigree, identification of a mutation in TGFB1, and review. Am J Med Genet A. 2004;129A(3):235-247.
doi pubmed - Bas F, Darendeliler F, Petorak I, Sadikoglu B, Bilir A, Bundak R, Saka N, et al. Deflazacort treatment in progressive diaphyseal dysplasia (Camurati-Engelmann disease). J Paediatr Child Health. 1999;35(4):401-405.
doi pubmed - Ayyavoo A, Derraik JG, Cutfield WS, Hofman PL. Elimination of pain and improvement of exercise capacity in Camurati-Engelmann disease with losartan. J Clin Endocrinol Metab. 2014;99(11):3978-3982.
doi pubmed - Balsan S, Steru D, Bourdeau A, Grimberg R, Lenoir G. Effects of long-term maintenance therapy with a new glucocorticoid, deflazacort, on mineral metabolism and statural growth. Calcif Tissue Int. 1987;40(6):303-309.
doi pubmed - Kim YM, Kang E, Choi JH, Kim GH, Yoo HW, Lee BH. Clinical characteristics and treatment outcomes in Camurati-Engelmann disease: A case series. Medicine (Baltimore). 2018;97(14):e0309.
doi pubmed - Moreira S, Cunha B, Jesus NP, Santos L. Pain improvement in Camurati-Engelmann disease after anti-TNFalpha therapy. BMJ Case Rep. 2017;2017.
doi pubmed - Combier A, Palazzo E, Forien M, Gardette A, Dieude P, Ottaviani S. Failure of conventional treatment and losartan in Camurati-Engelmann disease: A case report. Joint Bone Spine. 2018;85(5):649-650.
doi pubmed - Wahba Y, Abdel Ghaffar NA, Shaltout A, Elsharkawy A. Camurati-Engelmann disease: New clinical insights in an Egyptian case report. J Orthop Sci. 2020;25(3):529-532.
doi pubmed - Simsek-Kiper PO, Dikoglu E, Campos-Xavier B, Utine GE, Bonafe L, Unger S, Boduroglu K, et al. Positive effects of an angiotensin II type 1 receptor antagonist in Camurati-Engelmann disease: a single case observation. Am J Med Genet A. 2014;164A(10):2667-2671.
doi pubmed - Baroncelli GI, Ferretti E, Pini CM, Toschi B, Consolini R, Bertelloni S. Significant improvement of clinical symptoms, bone lesions, and bone turnover after long-term zoledronic acid treatment in patients with a severe form of Camurati-Engelmann disease. Mol Syndromol. 2017;8(6):294-302.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.