

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 9, Number 1, March 2020, pages 1-3
Arm Swelling From Subclavian Vein Thrombosis: Factor V Leiden Mutation
Shawn X. Wena, Katherine N. Tomb, Yousef Etoomc, d, e, Jonathan P. Wongc, Rosemary G. Moodiec, Ibrahim Al-Hashmic, Leonardo R. Brandaoc, e, Peter D. Wongc, e, f
aFaculty of Medicine, University of Toronto, Toronto, Canada
bFaculty of Health Sciences, McMaster University, Hamilton, Canada
cDepartment of Paediatrics, Hospital for Sick Children, Faculty of Medicine, University of Toronto, Toronto, Canada
dDepartment of Paediatrics, St. Joseph’s Health Centre, Toronto, Canada
eSickKids Research Institute, Toronto, Canada
fCorresponding Author: Peter Wong, Department of Paediatrics, Hospital for Sick Children, Faculty of Medicine, University of Toronto, 722-115 Humber College Blvd., Toronto, Ontario M9V 0A9, Canada
Manuscript submitted February 18, 2020, accepted February 28, 2020
Short title: Deep Vein Thrombosis From FVL Mutation
doi: https://doi.org/10.14740/ijcp343
| Abstract | ▴Top |
Factor V Leiden is a genetic condition that can decrease natural anticoagulant effect in response to activated protein C, increasing thromboembolic risk. We report a 17-year-old man, homozygous for the Factor V Leiden mutation, with sudden onset arm stiffness, pain and swelling caused by a deep vein thrombosis in the subclavian vein. Pediatricians should consider thromboembolic diseases in cases of spontaneous acute limb discomfort and discoloration with no obvious cause.
Keywords: Thromboembolic disease; Factor V Leiden; Deep venous thrombosis
| Introduction | ▴Top |
Factor V Leiden (FVL) is an autosomal dominant condition that causes poor natural anticoagulant response in the presence of activated protein C, therefore increasing the risk for venous thromboembolism. Of all individuals with the FVL mutation, only 1% are homozygous for the disorder and its prevalence depends on ethnicity [1]. We present a 17-year-old man who had sudden onset arm stiffness, pain and swelling caused by a deep vein thrombosis in the subclavian vein. A thrombophilia screening later determined the individual to be homozygous for the FVL mutation. Our case demonstrates that thromboembolic disease should be considered in the presentation of acute limb discomfort and discoloration, especially in the absence of trauma, compression syndromes or vascular or bony anomalies.
| Case Report | ▴Top |
A previously healthy 17-year-old man presented to his pediatrician with a 1-day history of arm stiffness, swelling and redness. He was a competitive athlete who while stretching at early morning practice noticed stiffness of the left shoulder which was followed by swelling and redness of the left upper limb. His symptoms progressed throughout the day despite treatment with acetaminophen. There was no history of trauma, immobilization or previous similar episode. He denied any constitutional, cardiac or respiratory symptoms.
The patient’s past history was not remarkable. He did not use prescription or use over-the-counter medication. He denied smoking, binge drinking alcohol or using illicit drugs. His parents were non-consanguineous. His father was of European descent; however, his mother was adopted and unaware of her biological family history. His older brother was healthy. There was no family history of thromboembolic disease.
On physical exam, he was well perfused and afebrile. His blood pressure (BP) was 120/70 mm Hg and heart rate was 60 beats per minute (bpm) and regular. He was breathing comfortably with normal oxygen saturation in room air. His left arm was dark purple in color and moderately swollen compared to the right. He had full range of motion of both shoulders and elbows. The circumference of the left upper arm was 1 cm, greater than the right. His peripheral pulses were good. There was no lymphadenopathy in the head, neck or axilla regions. His cardiac, respiratory and neurological examinations were unremarkable.
His complete blood count (hemoglobin (HGB) 157 g/L; white blood cells (WBCs) 7.9 × 109/L; platelet count 236 × 109/L) and renal function tests (urea 8.1 mg/dL; creatinine 84 umol/L) were normal. His partial thromboplastin time was 27.8 s and international normalized ratio (INR) was 1.1. An ultrasound examination and contrast venogram (Fig. 1) of the left shoulder confirmed a left subclavian deep vein thrombosis. A computerized tomography (CT) scan with contrast (i.e. CT venography) ruled out anatomical and vascular causes. A thrombophilia screening revealed the diagnosis.
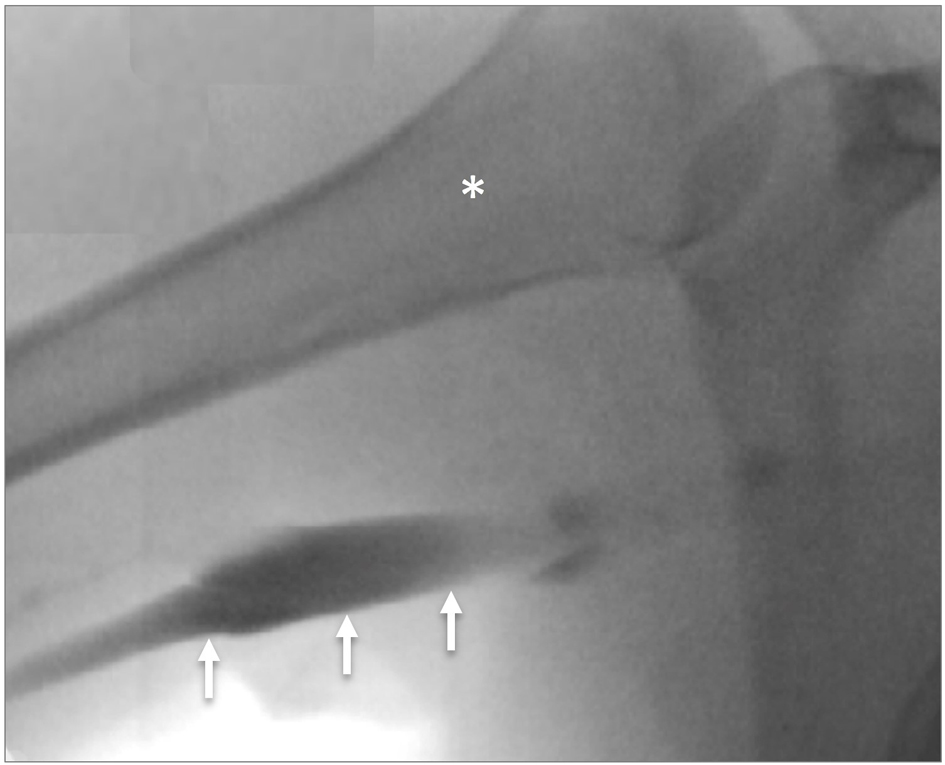 Click for large image | Figure 1. Contrast venogram demonstrating left subclavian vein thrombosis with occlusion (arrows). *Humerus. |
Thrombophilia screening identified the patient to be homozygous for FVL mutation and heterozygote for the methylene tetrahydrofolate reductase variant with normal homocysteine level. The remainder of the workup was negative for protein C, S and antithrombin, lipoprotein (a), prothrombin gene, lupus anticoagulant immunoglobulin G (IgG) antibodies, anticardiolipin, Factor VII, IX, and XI levels, and homocysteine levels. He was treated with low molecular weight heparin (200 IU/kg subcutaneously daily) followed by oral anticoagulation therapy (dosage to maintain target INR 2.5). He gradually returned to competitive sports with the occasional complaint of discomfort in his left upper limb. Over the next 2 years on anticoagulation therapy, his course remained uneventful. There was no recurrence of thromboembolic disease or post-phlebitic syndrome.
| Discussion | ▴Top |
Our case describes the first thrombotic event in the upper extremity of an adolescent man with homozygous FVL. The presentation at a young age before 30 years of age is unusual. Further, deep vein thromboses are more common in the lower limbs as compare to the upper limbs. Other unusual locations include cerebral, retinal, hepatic and renal veins. Homozygous FVL may also be associated with superficial vein thromboses. Finally, our case was absent for predisposing factors such as central venous catheter use, travel, injury, obesity or surgery.
FVL is a genetic disorder characterized by a poor natural anticoagulant effect from activated protein C and an increased risk for venous thromboembolism. Factor V is a procoagulant factor activated by thrombin that, in a positive feedback loop, increases thrombin production and subsequent activation of the clotting cascade. FVL is a single point mutation in the Factor V gene F5, which replaces arginine with glutamine at amino acid position 506. This mutation results in resistance in degradation of activated and inactivated Factor V by activated protein C. Given this is a natural negative feedback loop that controls excessive hemostatic responses, its derangement leads to a hypercoagulable state.
An autosomal dominant condition, FVL is the most common form of inherited thrombophilia [2, 3]. Its prevalence is highest in Europeans and rare in Asians and Africans [1]. Most FVL individuals are heterozygotes, while less than 1% are homozygotes. The relative risk for having a venous thromboembolism is 3 - 8 folds in heterozygotes and 10 - 80 times in homozygotes as compared to those without the mutation [4, 5]. Additionally, recurrence of venous thromboembolism risk is higher in homozygous forms [6, 7].
There are no specific clinical features of FVL deficiency. The major clinical manifestation of FVL is venous thromboembolism, such as pulmonary embolism and deep vein thrombosis, which may also occur in unusual locations. Symptomatic affected individuals are in adulthood with few before 30 years of age [4]. Further, the individual’s thrombotic risk is dependent on their zygosity and presence of other genetic or acquired thrombophilia disorders. Hormonal shifts such as use of oral contraceptives, hormone replacement therapy or pregnancy can compound risk of thrombosis secondary to FVL mutation alone. Further, clinical risk may be affected by obesity, air travel, malignancy or trauma. However, heterozygosity for FVL deficiency is not associated with an increase in mortality or reduced life expectancy [8, 9].
The mainstay of FVL carriers or individuals affected by FVL homozygosity who develop an acute venous thrombotic event is anticoagulation therapy. Heterozygous forms of FVL generally do not require long-term anticoagulation [1]. Conversely, longer anticoagulation therapy may be considered for affected individuals with first or recurrent unprovoked venous thromboembolism, presence of additional thrombophilia disorders. The literature on length of anticoagulation in children is scarce and such decisions should also involve patient/family preferences.
Conclusions
We describe a male athlete with arm pain, swelling and blueness from an unprovoked deep vein thrombosis of the subclavian vein. Thrombophilia screening identified the homozygous FVL mutation. It is an autosomal dominant condition and most common form of inherited thrombophilia, but has a higher risk of developing venous thromboembolism, compared to heterozygous forms. Long-term anti-coagulation therapy may be indicated. Although there are no diagnostic clinical features of FVL, unprovoked deep vein thrombosis or pulmonary embolus may be first presentations. Therefore, thromboembolic disease-causing acute limb discomfort and discoloration should be considered in the absence of trauma, compression syndromes or vascular or bony anomalies.
Acknowledgments
We acknowledge the patient and family for consenting for information and images to be published.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Family consent has been obtained in writing.
Author Contributions
SXW, KNT, and JPW wrote the first draft. LRB and IA-H provided expert review. Senior authors were RGM, PDW, and YE. All authors provided the clinical case concept and reviewed the final manuscript.
Data Availability
The authors declare that data supporting the findings of the study are available within the article.
| References | ▴Top |
- Kujovich JL. Factor V Leiden thrombophilia. Genet Med. 2011;13(1):1-16.
doi pubmed - Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg PR, Miletich JP. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med. 1995;332(14):912-917.
doi pubmed - Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood. 1995;85(6):1504-1508.
doi pubmed - Linnemann B, Meister F, Schwonberg J, Schindewolf M, Zgouras D, Lindhoff-Last E, registry M. Hereditary and acquired thrombophilia in patients with upper extremity deep-vein thrombosis. Results from the MAISTHRO registry. Thromb Haemost. 2008;100(3):440-446.
doi pubmed - Martinelli I, Battaglioli T, Bucciarelli P, Passamonti SM, Mannucci PM. Risk factors and recurrence rate of primary deep vein thrombosis of the upper extremities. Circulation. 2004;110(5):566-570.
doi pubmed - Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med. 2006;166(7):729-736.
doi pubmed - Segal JB, Brotman DJ, Necochea AJ, Emadi A, Samal L, Wilson LM, Crim MT, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA. 2009;301(23):2472-2485.
doi pubmed - Hille ET, Westendorp RG, Vandenbroucke JP, Rosendaal FR. Mortality and causes of death in families with the factor V Leiden mutation (resistance to activated protein C). Blood. 1997;89(6):1963-1967.
doi pubmed - Heijmans BT, Westendorp RG, Knook DL, Kluft C, Slagboom PE. The risk of mortality and the factor V Leiden mutation in a population-based cohort. Thromb Haemost. 1998;80(4):607-609.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.