

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 3, Number 1, March 2014, pages 16-18
Extrahepatic Biliary Atresia With Cholestatic Jaundice and Hydrocephalus
Vijay Baburao Sonawanea, b, Rahul Malhotraa, Sadhana Purandarea
aDepartment of Pediatrics, Dr. D.Y. Patil Medical College, Navi Mumbai, India
bCorresponding author: Vijay Baburao Sonawane, Department of Pediatrics, Dr. D.Y. Patil Medical College and Hospital, Navi Mumbai, Maharashtra, India
Manuscript accepted for publication February 20, 2014
Short title: Extrahepatic Biliary Atresia
doi: https://doi.org/10.14740/ijcp137w
| Abstract | ▴Top |
Biliary atresia is the most frequent cause of surgical jaundice in the late neonatal age group. Congenital extrahepatic biliary atresia and obstructive hydrocephalus is a rare association and not reported earlier in literature. We report a 2-month-old baby who presented with yellow discoloration of skin, high colored urine and clay colored stools since 20 days of life. Head size was apparently looking large with dilated veins over the scalp. The baby was diagnosed and treated as extrahepatic biliary atresia that led to cholestatic jaundice and cerebral aqueductal stenosis with hydrocephalus.
Keywords: Extrahepatic biliary atresia; Cerebral aqueductal stenosis; Cholestatic jaundice and hydrocephalus
| Introduction | ▴Top |
Biliary atresia is characterized by a biliary obstruction of unknown origin that presents in the neonatal period. It is the most frequent cause of surgical jaundice in the late neonatal age group. Depending on the level of obstruction, biliary atresia is classified into three types. Type 1 was common bile duct obstruction, type 2 was obstruction at the level of hepatic duct and type 3 was obstruction of the porta hepatis.
We report a 2-month-old baby with congenital extrahepatic biliary atresia and obstructive hydrocephalus which is a rare association and not reported earlier in literature.
| Case Report | ▴Top |
A 2-month-old male baby presented with yellow discoloration of skin, gradual in onset, progressive with passage of high colored urine and clay colored stools since 20 days of life. Antenatally, mother had preeclampsia. The baby was second of the twin, born at term, by LSCS, did not cry immediately after birth requiring resuscitation, weighed 1800 g and was admitted in NICU for 1 month. Baby had hypoglycemic convulsion on the second day of life and also necrotizing enterocolitis which was managed conservatively, given 3 units of blood transfusion. The other twin is normal. Developmentally, the baby does not recognize mother, hands are mostly closed and social smile is not achieved.
On physical examination, baby was marasmic, sick looking, irritable and afebrile with stable vital parameters. Length, weight and head circumference of the child were 47 cm, 1800 g and 37 cm respectively. Baby had triangular facies, severe anemia and deep jaundice. Head size was apparently looking large with dilated veins over the scalp. Anterior fontanel was wide open, pulsatile and diamond shaped with marked sutural separation. Posterior fontanel was patent.
On CNS examination, baby was alert and active, pupils were reacting to light bilaterally but not following light, ophthalmic evaluation revealed grade 4 retinopathy of prematurity and baby also had hypertonia and hyperreflexia. Ventral suspension showed, head was momentarily in the same plane of back. Baby had hepatosplenomegaly, liver 5 cm enlarged with 8 cm span, smooth surface, firm in consistency and well-defined margins associated with mild splenomegaly (1.5 cm in size).
Hematological workup revealed anemia (9.1 g/dL), leukocytosis (29.3 k/µL) and thrombocytosis (782 k/µL). Blood culture showed growth for streptococcus species and liver function tests revealed a total bilirubin of 11.6 mg/dL with direct and indirect bilirubin of 7.2 mg/dL and 4.4 mg/dL, and SGPT and SGOT were 459 and 388 U/L respectively. Hydrocephalus due to cerebral aqueductal stenosis was seen on USG skull. MRI of brain revealed gross active obstructive hydrocephalus (Fig. 1). TORCH titers were negative. HIDA scan showed mildly impaired hepatocyte function, hepatomegaly and extrahepatic biliary atresia.
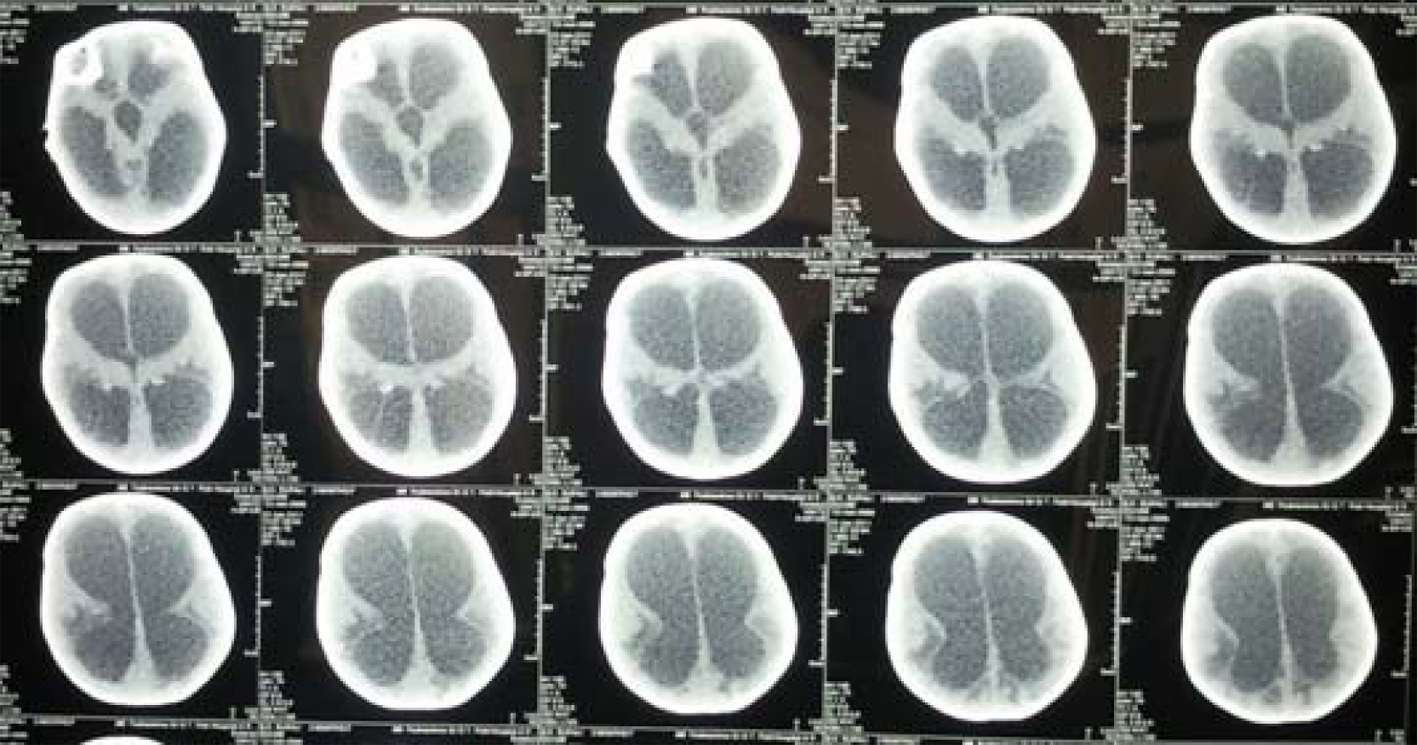 Click for large image | Figure 1. MRI of brain of the child showing gross active obstructive hydrocephalus. |
Baby was diagnosed as cholestatic jaundice due to extrahepatic biliary atresia with hydrocephalus due to aqueductal stenosis, sepsis with failure to thrive and anemia.
| Discussion | ▴Top |
Biliary atresia is a disease of unknown etiology characterized by a biliary obstruction of unknown origin that presents in the neonatal period [1]. It is the single, most frequent cause of death from liver disease and indication for liver transplantation (50% of all cases) in children [2]. Untreated, this condition leads to death in the first few years of life. It is not known to be a hereditary condition. The reported incidence of this disease varies from 5/100,000 to 32/100,000 live births, and is highest in Asia and Pacific regions [3].
The hepatic architecture is well preserved early in the course of the disease. Bile plugs in the ducts of portal triads indicate biliary obstruction. Canalicular and cellular bile stasis reflect the severity of the cholestasis [4].
Two different forms of biliary atresia have been identified [5]. One is syndromic biliary atresia (10%) associated with various congenital anomalies such as polyslpenia, asplenia, situs inversus, pre-duodenal portal vein, absence of retro-hepatic inferior vena cava and intestinal malrotation. The other is non-syndromic biliary atresia (90%), an isolated anomaly. The reported incidence of malformation among the children affected with extrahepatic biliary atresia is cardiovascular (60%), digestive (47%), genitourinary (13%) and splenic anomalies (40%) [6]. The prognosis of untreated biliary atresia is extremely poor with death from liver failure usually occurring within 2 years [7]. However, the hepatoportoenterostomy procedure can restore bile flow in most infants who undergo the procedure, but is rarely curative.
Our patient had extra hepatic biliary atresia, associated with CNS anomaly in the form of obstructive hydrocephalus which has not been reported earlier. Our patient also had retinopathy of prematurity. He was treated conservatively and referred to higher center for surgical intervention.
Themis et al in their study of 237 patients with extrahepatic biliary atresia found that 20% had associated congenital anomalies, and of these 28 had cardiovascular, 22 digestive and 19 splenic malformations. Hirofumi et al in a study on 87 patients with biliary atresia reported associated anomalies in seven (8%). These anomalies included situs inversus in four, polysplenia in five, preduodenal portal vein in five, absent portal vein in one, absent inferior vena cava in two, malrotation of the intestine in five and congenital heart disease in three patients [8].
| References | ▴Top |
- Alagille D. Extrahepatic biliary atresia. Hepatology. 1984;4(1 Suppl):7S-10S.
doi pubmed - Whitington PF, Balistreri WF. Liver transplantation in pediatrics: indications, contraindications, and pretransplant management. J Pediatr. 1991;118(2):169-177.
doi - Biliary atresia. Author: Christophe Chardot.
- Desmet VJ. Cholangiopathies: past, present, and future. Semin Liver Dis. 1987;7(2):67-76.
doi pubmed - AHoward ER, editors Pediatric surgery and Urology: Long term outcomes. London: WB Saunders; 1998. P. 402-416.
- Silveira TR, Salzano FM, Howard ER, Mowat AP. Congenital structural abnormalities in biliary atresia: evidence for etiopathogenic heterogeneity and therapeutic implications. Acta Paediatr Scand. 1991;80(12):1192-1199.
doi pubmed - Kobayashi A, Itabashi F, Ohbe Y. Long-term prognosis in biliary atresia after hepatic portoenterostomy: analysis of 35 patients who survived beyond 5 years of age. J Pediatr. 1984;105(2):243-246.
doi - Biliary atresia associated with congenital structural anomalies. Hirofumi Tanano, Toshimichi Hasegawa, Hisayoshi Kawahara, Takashi Sasaki, Akira Okada.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.