

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website https://www.theijcp.org |
Case Report
Volume 13, Number 2, June 2024, pages 60-65

New Variant Found in the Bone Morphogenetic Protein Receptor 1B Gene in a Neonate With Trisomy 21
Mahmoud Galal Ahmeda, b, Muzammil Hafeeza, b, e, Faatimah Irfaanah Muzammilc, Anwar Khana, b, Hisham Hamdand, Mostafa Abdel Raouf El Bolkinia, Faatimah Maryam Muzammilb
aDepartment of Neonatology, Dubai Hospital, Dubai Health, Dubai, UAE
bDepartment of Pediatrics, Dubai Medical College, Dubai, UAE
cDepartment of Pediatrics, Al Jalila Children’s Specialty Hospital, Dubai Health, Dubai, UAE
dDepartment of Pediatric Pulmonology and Sleep Medicine, Al Jalila Children’s Specialty Hospital, Dubai Health, Dubai, UAE
eCorresponding Author: Muzammil Hafeez, Dubai Hospital, Dubai Health, Dubai, UAE
Manuscript submitted March 6, 2024, accepted May 6, 2024, published online June 23, 2024
Short title: New Variant in BMPR1B Gene in a Neonate
doi: https://doi.org/10.14740/ijcp534
| Abstract | ▴Top |
We present a case of a 29-week preterm infant delivered by emergency cesarean section due to suspected placental abruption, with a birth weight of 1.17 kg. The APGAR score was 7 at 1 min and 9 at 5 min. The baby was transferred on non-invasive ventilator support to the neonatal intensive care unit (NICU). Physical examination revealed features consistent with Down syndrome. Subsequently, he developed worsening respiratory distress, necessitating intubation and mechanical ventilation, ultimately requiring high-frequency oscillatory ventilation (HFOV). Despite surfactant administration, there was no improvement in the respiratory status. Echocardiography revealed severe pulmonary hypertension. Treatment with nitric oxide and later sildenafil was initiated, but there was limited clinical improvement. After consultation with a pediatric pulmonologist, genetic testing was pursued to investigate the possibility of mutations in surfactant-related genes. The genetic analysis identified a novel missense variant (p.His391Arg) in the bone morphogenetic protein receptor 1B (BMPR1B) NM_001203.3 gene, which has not been previously reported in disease-associated contexts and is not present in large population databases such as the Genome Aggregation Database (gnomAD) and the Greater Middle East (GME) variome database. Unfortunately, despite intensive care efforts, the infant remained critically ill and passed away on day 43 of life. Through this case report, we aim to highlight this novel mutation in the BMPR1B gene for pediatricians and neonatologists worldwide. We also seek to raise awareness about the associated clinical features and the importance of considering such mutations in the differential diagnosis. Furthermore, we advocate for ongoing research to develop improved management strategies that may enhance survival outcomes for infants with similar genetic mutations.
Keywords: Neonatology; Pulmonary arterial hypertension; New variant in BMPR1B gene; NICU; Mutation; Ventilation; Acromesomelic dysplasia; Brachydactyly; Bone morphogenetic proteins; Nitric oxide
| Introduction | ▴Top |
Pulmonary arterial hypertension (PAH) is an uncommon condition marked by remodeling of pulmonary arterioles, increased arterial pressure, resistance, and eventual heart failure. Pediatric-onset PAH, in contrast to its adult counterpart, displays greater variability and is frequently linked to a more unfavorable prognosis. The condition exhibits etiological diversity, encompassing familial PAH (FPAH), sporadic/idiopathic PAH (IPAH), and hereditary PAH (HPAH, comprising FPAH and IPAH with identified mutations) [1].
Pediatric PAH includes IPAH, HPAH, and PAH associated with congenital heart disease (CHD) and with abnormal lung development [2].
While the genetics of PAH in children have not undergone extensive exploration, available data indicate a potential divergence in genetic etiology between pediatric and adult PAH. Limited sample sizes in pediatric studies underscore the need for larger, more comprehensive investigations.
Reported mutations in the bone morphogenetic protein receptor type 2 (BMPR2) gene, activin receptor-like kinase 1 (ALK1) gene, and SMAD8 gene are associated with both HPAH and IPAH [3].
Bone morphogenetic proteins (BMPs) belong to the tumor necrosis factor-beta (TNF-β) family and play important roles in morphogenesis. BMPs are active during early development and their function is important in the neurological, cardiovascular, gastrointestinal, urinary, adipose, and musculoskeletal systems; thus, these proteins were proposed to be called body morphogenic proteins [4].
The BMP receptors (BMPRs) have been classified in two groups: type I, containing the ALKs, and type II, containing three distinct receptors: BMPR-II, ACTR-II, and ACTR-IIB. The type I receptors are subdivided into three sub-groups: the BMPR1 and ALK1 groups, activating Smad1/5/8 proteins and the TβR-I group, which interacts with SMAD2/3 [5]. The BMPR1 group comprises the BMPR-IA and BMPR-IB receptors; though other receptors are expressed in various cell types, the expression of BMPR-IB, encoded by the bone morphogenetic protein receptor 1B (BMPR1B) gene, is significant in the adrenal, brain, endometrial, ovarian and prostate tissue [4].
Along with PAH, acromesomelic dysplasia and brachydactyly have also been seen in significant mutations with the BMPR1B gene mutation.
In a recent study involving 88 neonates, no differences were observed in the majority of candidate genes, such as BMPR2 and nitric oxide (NO) synthase. However, a notable association with genetic variants related to cortisol signaling (specifically corticotropin-releasing hormone receptor-1 (CRHR1) and CRH-binding protein) was identified in cases of persistent pulmonary hypertension of the newborn (PPHN). Additionally, elevated levels of 17-hydroxyprogesterone were noted in infants with PPHN [6].
We hereby present a case of trisomy 21 with a mutation in the BMPR1B, causing a new heterozygous pathogenic variant, which gene has not been previously reported in individuals with disease and is absent from the large population studies such as the Genome Aggregation Database (gnomAD) and the Greater Middle East (GME) variome database.
| Case Report | ▴Top |
A baby was born to 41-year-old, gravida 2 para 1, at 29 weeks of gestation. Mother was an un-booked patient in our facility but following in another facility.
The baby was conceived spontaneously to a non-consanguineous couple as the first child of her second marriage. The mother had no previous medical history or known allergies. In her previous pregnancy (from her first marriage), she underwent a cesarean section due to failed induction of labor.
During her current pregnancy, the mother did not undergo an oral glucose tolerance test (OGTT) or group B streptococcus (GBS) test. However, her non-invasive prenatal testing (NIPT) indicated a high suspicion of trisomy 21.
Antenatal scans done showed normal amniotic fluid with fetus measuring appropriate for gestational age.
The mother initially sought care at another facility with complaints of premature leaking (clear and non-bloody), which was not accompanied by abdominal pain. She received her first dose of betamethasone before being transferred to our facility.
Baby was born by emergency cesarean section with suspicion of placental abruption at 29 weeks of gestation with a birth weight of 1.17 kg. The baby cried immediately after birth, but had shallow respiration and required positive pressure ventilation. APGAR score was 7 at 1 min and 9 at 5 min. The baby’s vitals showed bradycardia (heart rate (HR) < 100) at birth, which improved to HR of 140 with positive pressure T-piece ventilation. The respiratory rate was 40/min with mild subcostal recession, temperature was 36 °C and oxygen saturation during resuscitation ranged from 85% to 95%.
On initial examination after birth, child was initially active and had flat anterior fontanelle. He had features of Downs syndrome (up slanting of both eyes, depressed nasal bridge, low set ears, unilateral simian crease, wide spaced nipples and rocker bottom feet). The skin was warm and had a good turgor. The capillary refill time was less than 2 s.
He had normal heart rate, rhythm and normal pulses. No murmur was appreciated. He had tachypnea with mild subcostal recessions. His abdomen was soft and flat. He was active and no motor neurological deficit.
After initial assessment, he started having desaturation and bradycardia and was immediately connected to non-invasive mode of ventilation (positive end-expiratory pressure (PEEP) 6, pressure 20, rate 30/min and FIO2 of 30%). Baby stabilized and was transferred to neonatal intensive care unit (NICU).
In the NICU, the baby was vitally stable with HR of 140, and saturation of 97% with a blood pressure (BP) of 65/33 mm Hg and mean arterial pressure (MAP) of 35 mm Hg. His respiratory distress worsened and surfactant was administered using the less invasive surfactant administration (LISA) method, and the baby was kept on non-invasive ventilation. Despite the surfactant administration, the respiratory distress worsened, necessitating intubation and mechanical ventilation. A chest X-ray revealed slightly reduced lung volumes with diffusely increased broncho-vascular and interstitial markings, which were likely suggestive of respiratory distress syndrome, as shown in Figure 1. His FiO2 requirement increased to 90%.
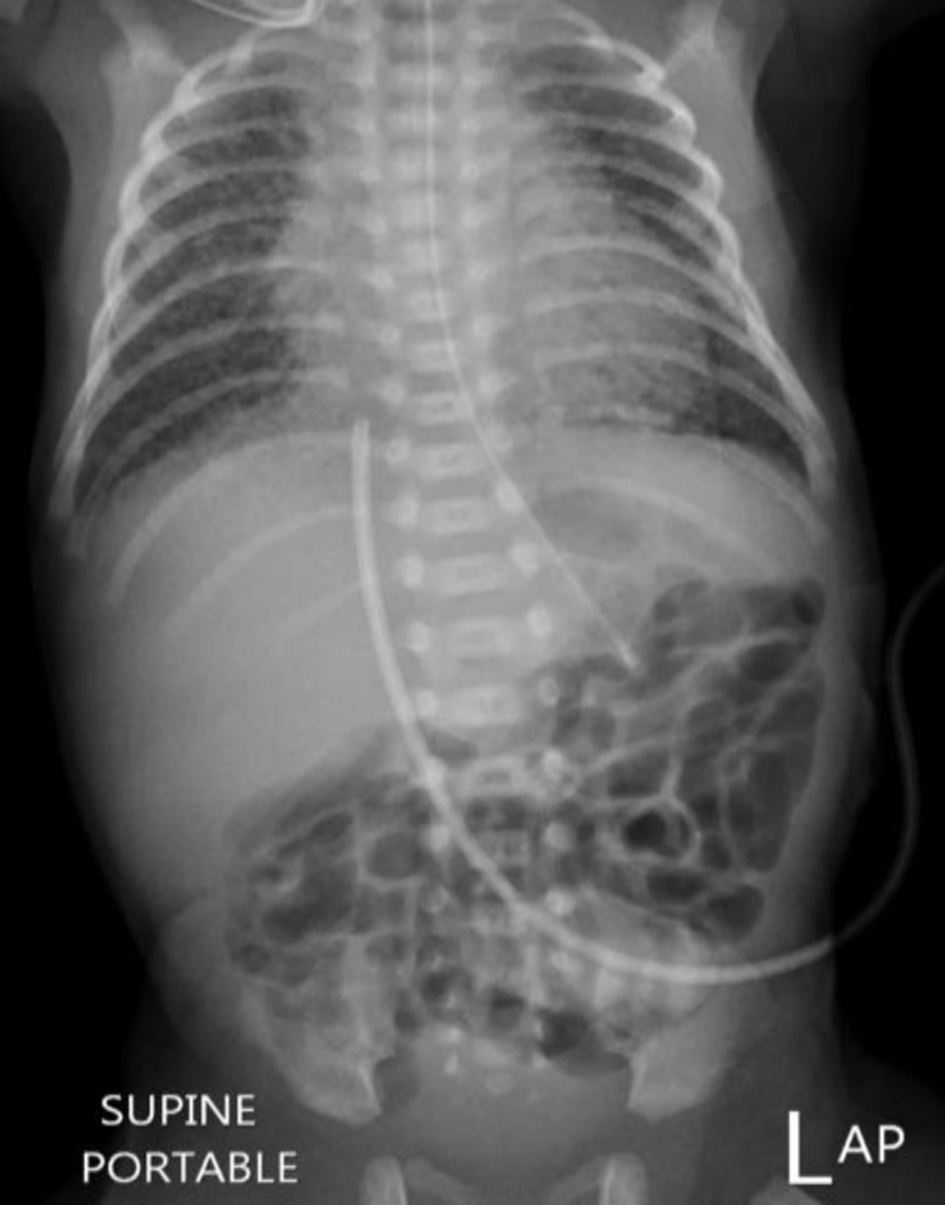 Click for large image | Figure 1. Chest X-ray after birth. |
Second dose of surfactant was administered but there was no significant clinical improvement. He continued to have desaturation and required high ventilator settings (Fig. 2).
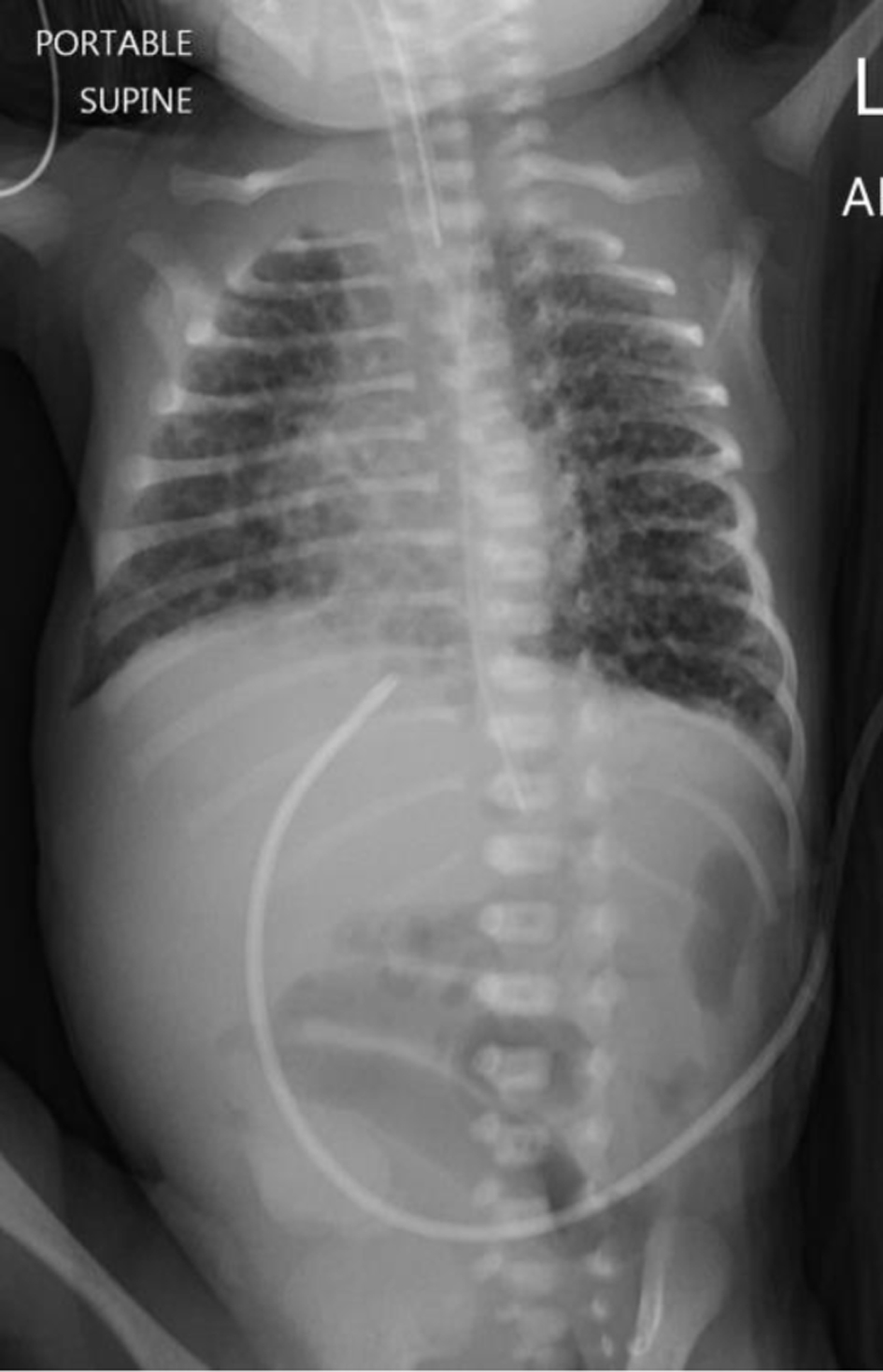 Click for large image | Figure 2. Chest X-ray after admission after two doses of surfactant. Features suggest the possibility of pulmonary interstitial emphysema in a known respiratory distress. |
Initial echo showed moderate pulmonary hypertension with large patent ductus arteriosus (PDA) (3.6 mm with left to right shunt).
Trial of high-frequency ventilation was given; however, the baby did not tolerate high-frequency ventilation and was shifted back to assist control mode of ventilation. The baby continued to have desaturations and there was difference of more than 10% in pre- and post-ductal saturation. NO was started in view of the pulmonary hypertension and on day 28 of life, the dose reached 35 ppm. He was also started on sildenafil which showed transient improvement in his oxygen saturation; however, he again started to have severe desaturations.
Vasopressin was started. Baby developed severe hyponatremia, hence had to be stopped immediately. High ventilator settings were continued for the baby. Repeat echo done on day 28 of life while he was on NO showed pulmonary hypertension, large PDA (with right to left shunt), atrial septal defect secundum (ASD) II, so there was shunt reversal.
Repeat chest X-ray was done in view of clinical worsening, showing severe pulmonary interstitial emphysema (PIE) picture, honey-comb appearance and cystic changes as shown in Figure 3.
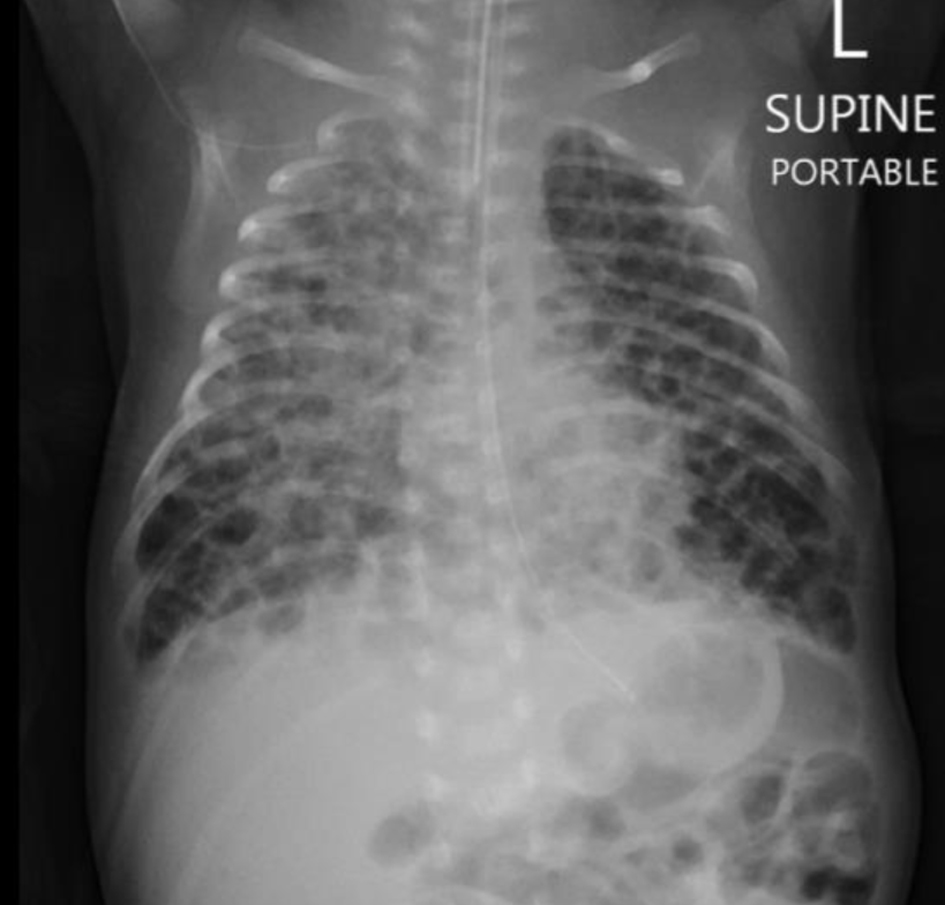 Click for large image | Figure 3. Chest X-ray showing severe pulmonary interstitial emphysema picture, honey-comb appearance and cystic changes. |
A karyotype was performed and cytogenetics study by karyotyping showed an abnormal male chromosome complement with a non-disjunction trisomy of chromosome 21 in all the metaphase cells studied.
The opinion of the pulmonologist was taken, who advised us to conduct genetic tests considering the possibility of a mutation in the surfactant genes. Subsequently, the genetic test results revealed a novel variant p.(His391Arg) in the BMPR1B gene, as shown in Table 1. His other investigations including blood counts, creatinine, urea and liver function tests were normal. However, his CRP was positive. There was no growth seen in blood and respiratory culture, as shown in Table 2.
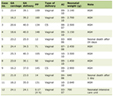 Click to view | Table 1. Genetic Test Result (Custom Gene Panel) |
Click to view | Table 2. Lab Investigations |
Genetic test official report with impression
Sequencing of the coding regions and splice sites of the 58 genes associated with the condition identified a heterozygous variant of uncertain significance in BMPR1B. In variant details, it was mentioned that the p.His391Arg missense variant in the BMPR1B gene has not been previously reported in individuals with disease and is absent from the large population studies such as the gnomAD and the GME variome database.
Initial brain ultrasound (US) done soon after admission was normal. Follow-up brain US done later showed persistent periventricular flaring and otherwise there was no evidence of any intracranial bleed or hydrocephalus.
The baby in our case remained critical and unfortunately passed away on day 43 of life. No autopsy was performed.
| Discussion | ▴Top |
In summary, our case involves a neonate born at 29 weeks via emergency C-section and subsequently transferred to the NICU due to respiratory distress. The ventilation mode was upgraded from non-invasive to mechanical ventilation, with minimal improvement despite administering two doses of surfactant. An echocardiogram (ECHO) revealed a PDA initially exhibiting left-to-right shunting, which later reversed to right-to-left shunting. Persistent pulmonary hypertension ensued and progressed to a severe form despite treatment with NO and sildenafil.
Karyotyping confirmed trisomy 21, and a custom gene panel (whole exome sequencing) revealed a novel mutation, p.His391Arg missense variant, in the BMPR1B gene. Notably, this mutation has not been previously reported in individuals with the disease and is absent from large population databases such as the gnomAD and the GME variome database.
PPHN represents a severe complication associated with several neonatal respiratory disorders, leading to adverse outcomes such as asphyxia, chronic lung disease, neurodevelopmental sequelae, and mortality. Despite the application of advanced therapeutic approaches such as high-frequency oscillatory ventilation, inhaled NO, and extracorporeal membrane oxygenation, PPHN remains challenging [7, 8]. While primary causes include factors like chronic hypoxia, meconium aspiration syndrome, birth asphyxia, sepsis, infection, and respiratory distress syndrome, there is a growing acknowledgment of a genetic contribution to PPHN [8-10].
Our premature baby, born at 29 weeks of gestation, experienced escalating respiratory distress. Initially diagnosed with hyaline membrane disease based on X-ray findings indicating reduced lung volume and increased broncho-vascular markings, along with an increasing FiO2 requirement, the infant received four doses of surfactant. Despite these interventions, persistent desaturation and a high FiO2 requirement persisted. The pre- and post-ductal oxygen saturation exhibited a difference exceeding 10. Initial ECHO revealed moderate pulmonary hypertension. In response to ongoing desaturation and evidence of pulmonary hypertension, NO was initiated at 20 ppm, initially showing improvement in oxygen saturation, but the response was transient, with continued desaturations. Considering the possibility of genetic surfactant abnormalities, a genetic study was conducted, revealing a mutation in the BMPR1B gene.
PAH in BMPR1B mutation
Based on clinical classification, WHO group 1 PAH includes sporadic disease (IPAH), HPAH, and association with certain medical conditions (associated PAH) [11]. Variants identified in known PAH risk genes were classified as pathogenic, likely pathogenic, or of uncertain significance according to the American College of Medical Genetics and Genomics guidelines [1].
Familial cases of PAH have been long recognized, and are usually inherited as autosomal dominant traits with incomplete penetrance; females are preferentially affected. HPAH accounts for about 6% of PAH cases [1]. HPAH is inherited in an autosomal dominant fashion with 10-20% penetrance and affects females approximately twice as often as males [3].
A genetic predisposition can lead to the rare disease PAH. Most mutations have been identified in the gene BMPR2 in HPAH [12].
Mutations in ALK-1, ENG, SMAD4 and SMAD8, other TGF-β family members, are additional rare causes of PAH [8].
In an article published in 2012, in the Circulation Journal, the authors identified two novel mutations in BMPR1B in two patients with IPAH [3], two BMPR1B missense mutations in two independent probands with IPAH. In proband A, c.479 G>A p.S160N was identified. In proband B, c.1176 C>A p.F392L was identified [3].
In a study published in 2003, the authors found that angiopoietin-1 down-regulates steady-state levels of BMPR1A mRNA and protein in subcultured human pulmonary arteriolar endothelial cells. These results unite nonfamilial primary pulmonary hypertension and multiple forms of secondary pulmonary hypertension by demonstrating that they have a similar pattern of aberrant gene expression and suggest that pulmonary hypertension may occur through a molecular cascade whereby angiopoietin-1 ultimately down-regulates steady-state levels of BMPR1A [13].
Acromesomelic dysplasia in BMPR1B mutation
Acromesomelic chondrodysplasias (ACD) are characterized by shortening and malformation of the limbs, predominantly the forearms, forelegs, hands and feet, and generally manifest also with short stature. Inheritance is autosomal recessive. The majority of the cases result from growth/differentiation factor 5 (GDF5) variants, and four homozygous variants in BMPR1B also have been reported in ACD. The first such BMPR1B variant is severe and leads to acromesomelic dysplasia type Demirhan, similar to chondrodysplasia-type Grebe, caused by biallelic GDF5 variants [14]. Not homozygous but heterozygous BMPR1B variants are known to cause brachydactyly, either type A1 (BDA1D) or type A2 (BDA2) [14].
Brachydactyly in BMPR1B mutation
Heterozygous mutations in the BMPR1B gene have been identified in persons with hypoplasia/aplasia of their phalanges and disturbance of interdigital joint structures. BMPR1B mutations have been reported in three German families with brachydactyly A2 or brachydactyly C with symphalangism-like phenotypes [15].
In a study published in June 2015, the authors have detected a novel heterozygous mutation substituting arginine at position 486 of the BMPR1B gene product [15]. The presence of a mutation affecting the same amino acid in BMPR1B in this family, as well as in the previously reported nonrelated German families raises the possibility of a significant hotspot within the BMPR1B gene.Of diagnostic importance for persons presenting with BMPR1B-related brachydactyly is a distinct entity recognizable on a clinical and radiological basis [4].
Learning points
1) To learn that there is a new variant (novel) mutation (p.His391Arg) in the BMPR1B NM_001203.3 gene and to promote more research on this gene, its effects and possible management strategies for aiding in better and survival outcomes.
2) For any neonate with persistent PAH, not responding to potent vasodilators like NO and sildenafil, genetic tests should be done to rule out genetic mutations giving rise to the clinical condition.
3) Any respiratory distress in prematurity, not responding to surfactant and ventilation, rules out surfactant protein mutations.
4) The need to evaluate any additional genetic variants in established genetic syndromes and its unusual phenotypes, if any.
5) Handling a critical neonate can be very challenging but it is important to think in all directions to improve outcome, should have coordinated care among different relevant specialties of pediatrics and think outside the box for causes when even after following guidelines and protocol of treatment, the baby is not improving.
Acknowledgments
None to declare.
Financial Disclosure
Authors declare no support in form of grants, equipment or drugs.
Conflict of Interest
Authors report no conflict of interest.
Informed Consent
Informed consent has been obtained.
Author Contributions
All authors have contributed to the writing and proofreading of the manuscript. Faatimah Irfaanah Muzammil worked on the literature review and structure of the article. Faatimah Maryam Muzammil and Muzammil Hafeez worked on data collection for the case and formulating the tables. Mahmoud Galal Ahmed, Anwar Khan, Hisham Hamdan and Mostafa Abdel Raouf ElBolkini worked on extensive editing and proofreading of the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Zhu N, Gonzaga-Jauregui C, Welch CL, Ma L, Qi H, King AK, Krishnan U, et al. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ Genom Precis Med. 2018;11(4):e001887.
doi pubmed pmc - Ma L, Chung WK. The role of genetics in pulmonary arterial hypertension. J Pathol. 2017;241(2):273-280.
doi pubmed pmc - Chida A, Shintani M, Nakayama T, Furutani Y, Hayama E, Inai K, Saji T, et al. Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ J. 2012;76(6):1501-1508.
doi pubmed - Bednarek M, Trybus M, Kolanowska M, Koziej M, Kiec-Wilk B, Dobosz A, Kotlarek-Lysakowska M, et al. BMPR1B gene in brachydactyly type 2-A family with de novo R486W mutation and a disease phenotype. Mol Genet Genomic Med. 2021;9(3):e1594.
doi pubmed pmc - Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147(1):35-51.
doi pubmed - Steinhorn RH. Advances in neonatal pulmonary hypertension. Neonatology. 2016;109(4):334-344.
doi pubmed - Nakwan N, Jain S, Kumar K, Hosono S, Hammoud M, Elsayed YY, Ariff S, et al. An Asian multicenter retrospective study on persistent pulmonary hypertension of the newborn: incidence, etiology, diagnosis, treatment and outcome. J Matern Fetal Neonatal Med. 2020;33(12):2032-2037.
doi pubmed - Jain A, McNamara PJ. Persistent pulmonary hypertension of the newborn: advances in diagnosis and treatment. Semin Fetal Neonatal Med. 2015;20(4):262-271.
doi pubmed - Rocha G, Baptista MJ, Guimaraes H. Persistent pulmonary hypertension of non cardiac cause in a neonatal intensive care unit. Pulm Med. 2012;2012:818971.
doi pubmed pmc - Steurer MA, Jelliffe-Pawlowski LL, Baer RJ, Partridge JC, Rogers EE, Keller RL. Persistent pulmonary hypertension of the newborn in late preterm and term infants in California. Pediatrics. 2017;139(1):e20161165.
doi pubmed - Ma L, Chung WK. The genetic basis of pulmonary arterial hypertension. Hum Genet. 2014;133(5):471-479.
doi pubmed - Eichstaedt CA, Sassmannshausen Z, Shaukat M, Cao D, Xanthouli P, Gall H, Sommer N, et al. Gene panel diagnostics reveals new pathogenic variants in pulmonary arterial hypertension. Respir Res. 2022;23(1):74.
doi pubmed pmc - Du L, Sullivan CC, Chu D, Cho AJ, Kido M, Wolf PL, Yuan JX, et al. Signaling molecules in nonfamilial pulmonary hypertension. N Engl J Med. 2003;348(6):500-509.
doi pubmed - Yildirim Y, Ouriachi T, Woehlbier U, Ouahioune W, Balkan M, Malik S, Tolun A. Linked homozygous BMPR1B and PDHA2 variants in a consanguineous family with complex digit malformation and male infertility. Eur J Hum Genet. 2018;26(6):876-885.
doi pubmed pmc - Badura-Stronka M, Mroz D, Beighton P, Lukawiecki S, Wicher K, Latos-Bielenska A, Kozlowski K. Novel mutation in the BMPR1B gene (R486L) in a Polish family and further delineation of the phenotypic features of BMPR1B-related brachydactyly. Birth Defects Res A Clin Mol Teratol. 2015;103(6):567-572.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.