

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website https://www.theijcp.org |
Case Report
Volume 13, Number 1, May 2024, pages 26-33

X-Linked Lymphoproliferative Disease Type 2 With Activation of Hemophagocytic Lymphohistiocytosis Secondary to Epstein-Barr Virus Infection
Shahad Hani Alsheikha, c, Noor Alsheikha, Mona AlSalehb
aCollege of Medicine, Imam Abdulrahman Bin Faisal University, Dammam 34212, Saudi Arabia
bHematology and Oncology Unit, Department of Pediatrics, King Fahad University Hospital, Al Aqrabiyah, Al Khobar 34445, Saudi Arabia
cCorresponding Author: Shahad Hani Alshiekh, College of Medicine, Imam Abdulrahman Bin Faisal University, Dammam 34212, Saudi Arabia
Manuscript submitted September 16, 2023, accepted October 21, 2023, published online January xx, 2023
Short title: XLP-2 With Activation of HLH and EBV
doi: https://doi.org/10.14740/ijcp525
| Abstract | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) is a rare yet potentially life-threatening syndrome. It is best described as uncontrolled immune system activation leading to hemophagocytosis. Our case report aims to present a practical approach, discuss the challenges in the diagnosis and management of HLH, and to shed light on the X-linked lymphoproliferative disease type 2 (XLP-2) gene, STXBP2 gene (as a carrier), and Epstein-Barr virus (EBV) infection in relation to HLH. This is a 2-year-8-month-old Saudi boy, who presented to the emergency department with fever for more than 7 days, not associated with any other major complaints. During the first couple of days of admission, primary results showed hyperferritinemia, low fibrinogen, anemia, high inflammatory markers, high D-dimer, and increasing liver enzymes and triglycerides. The patient fulfilled the diagnostic criteria of HLH on the third day of admission. Further investigations revealed a genetic mutation in XLP-2 gene, STXBP2 gene (as a carrier) and EBV infection causing HLH. HLH-94 protocol was used for management. The patient had an excellent clinical outcome, unexpectedly, by only using dexamethasone, with complete remission and no recurrence up to date. HLH is not frequently encountered in daily practice. However, it can be fatal. This case report describes a unique case of XLP-2, STXBP2 carrier with extreme EBV infection and activation of HLH, who was successfully brought into remission by partial application of HLH-94 protocol and careful clinical and laboratory monitoring. The diagnosis and management of HLH can be challenging. However, a reasonable clinical judgment can improve the outcomes.
Keywords: Hemophagocytic lymphohistiocytosis; XLP-2; EBV
| Introduction | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) is a potentially fatal syndrome. It can be described as uncontrolled activation of cytotoxic T lymphocytes and natural killer (NK) cells. This will result in extensive cytokine release, also known as cytokine storm, leading to macrophage hyperactivation. This hyperactivation will result in hemophagocytosis and immune-mediated injury to many organs [1, 2]. The first case reported in the literature was in 1939 by Scott and Robb-Smith, first described as histiocytic medullary reticulosis [3]. HLH is a rare disease, with an incidence estimated to be 1:800,000 per year in Japan [4]. Another Swedish study estimated the incidence of primary HLH to be 0.12 - 0.15 per 100,000 children per year [5].
HLH can occur as primary, also called genetic HLH. This subtype usually affects children at an early age. However, it can be diagnosed at any age. Another HLH subtype is secondary or sporadic HLH. This subtype usually affects any age and is associated with acquired disorders such as infections, malignancies, rheumatological or immunological disorders. Nevertheless, it is noted that primary disease needs an acquired disorder that acts as a trigger [1, 4, 6, 7].
Clinical presentation of HLH can be wide-ranging. However, the known presentation includes fever, splenomegaly, hepatomegaly, cytopenia, respiratory distress, renal failure, and central nervous system (CNS) toxicity. In addition, liver is commonly involved and is discovered by high aspartate aminotransferase (AST) and alanine aminotransferase (ALT), which may reflect hepatitis or liver failure [1, 8]. HLH-2004 criteria (Fig. 1) are most commonly used to diagnose HLH, which include either 1) establishing a molecular diagnosis of HLH; or 2) fulfilling five out of eight criteria, which include fever, splenomegaly, cytopenias affecting ≥ 2 of 3 lineages, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis demonstrated in the bone marrow or spleen or lymph node, low or absent NK cell activity, ferritin ≥ 500 µg/L, and high soluble CD25 levels (sIL-2 receptor) ≥ 2,400 U/mL [1, 8]. However, the diagnosis is still challenging due to the nonspecific symptoms that can constitute many other differential diagnoses, the uncommonness of the disease, and the nature of the disease, as it is known to be rapidly progressing with rapid evolution in severity, which limits the time for practitioner and necessitate intervention at an early stage of investigations that need high clinical sense and reasonable clinical judgment to prioritize investigations over others and decide when it is time to provide therapeutic actions [6]. Our case report aims to discuss the challenges in the diagnosis and management of HLH, and to shed light on X-linked lymphoproliferative disease type 2 (XLP-2) gene, STXBP2 gene - as a carrier - and Epstein-Barr virus (EBV) infection in relation to HLH.
 Click for large image | Figure 1. HLH-2004 diagnostic criteria commonly used in HLH diagnosis. HLH: hemophagocytic lymphohistiocytosis; NK: natural killer. |
| Case Report | ▴Top |
A 2-year-8-month-old Saudi boy, not known to have medical illnesses, presented to the emergency department with a fever for more than 7 days, measured axillary, reaching up to 39 °C. The fever was intermittent, peaked almost every 4 h, and responded to acetaminophen.
His fever was associated with sweating, redness of the eyes, and vomiting with every feed; however, the vomiting was non-bloody, non-bilious and non-projectile. Moreover, the mother reported a history of weight loss of around 1 kg in 1 week and decreased activity and feeding. There was no history of abnormal movements, loss of consciousness, weakness, photophobia, cracked lips, skin rash, jaundice, pallor, joint pain, ear pain or discharge, chest pain, shortness of breath, cough, sore throat, runny nose, abdomen pain, constipation, diarrhea, or urinary symptoms. In addition, there was no history of recent travel, or ingestion of unpasteurized milk. His mother reported a history of previous mild self-limited fever episodes, occurring every 3 - 4 months, lasting for 1 day and then spontaneously resolving. Also, she reported a positive history of contact with several family members with coronavirus disease 2019 (COVID-19) infection around 2 - 3 weeks prior to presentation to the hospital.
He was a product of normal vaginal delivery, full term, with an uneventful and uncomplicated pregnancy and delivery. His vaccines are up to date. His development is appropriate for his age, with unremarkable past medical and surgical history. His parents are non-consanguineous, and he does not have any siblings. However, the mother was pregnant during the time of the patient’s presentation.
Prior to presentation to our hospital, the patient visited another health facility. Parents were advised for bone marrow aspiration and biopsy due to cytopenias, but they refused and wanted a second opinion.
His physical examination was significant for palpable liver 2 cm below costal margin, and four inguinal lymph nodes were palpable, measuring around 1 cm; the rest of the examination was unremarkable.
Our patient was admitted as a case of fever for further investigation. Upon admission, he underwent investigations (Table 1) and was started on intravenous (IV) fluid, ceftriaxone and acetaminophen when needed. Primary results have shown extreme hyperferritinemia, low fibrinogen, anemia, high inflammatory markers, transaminitis and high D-dimer. Thus, multiple differential diagnoses, such as HLH, macrophage activation syndrome (MAS), multisystem inflammatory syndrome in children (MIS-C), Kawasaki and others, were suggested and needed to be ruled out.
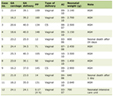 Click to view | Table 1. Significant Investigations Conducted Upon Admission |
During the first couple of days of admission, the patient still used to have multiple spikes of fever. However, he was stable hemodynamic wise. Follow-up of his investigations showed worsening of liver enzymes, with AST: 727, ALT: 208, lactate dehydrogenase (LDH): 4,970 and γ-glutamyl transpeptidase (γGTP): 285. The lipid profile showed a new increase in triglycerides at level of 386. Complete blood count (CBC) was dropping to the lower border with white blood cell (WBC): 6.4, red blood cell (RBC): 3.42, hemoglobin (Hb): 8.1 and platelet (Plt): 145. The erythrocyte sedimentation rate (ESR) rose to 32. Based on the lateral results, he was started on intravenous immunoglobulin (IVIG) and methylprednisolone (after obtaining the bone marrow samples), and ceftriaxone was switched to cefepime.
More investigations were conducted (Table 2). EBV early antigen (EA) immunoglobulin (Ig)G, EBV nuclear antigen (NA) IgG, EBV viral capsid antigen (VCA) IgG, EBV IgM, rubella IgG, varicella IgG, cytomegalovirus (CMV)-IgG, herpes simplex virus (HSV)-IgM, HSV-IgG, and COVID-19 IgG were positive. Urine protein was 29.3. Abdomen ultrasound (US) showed moderate hepatomegaly with biliary sludge. Results of bone marrow workup showed normocellular marrow for age with evident hemophagocytosis and no evidence of malignancy.
Click to view | Table 2. Additional Investigations Conducted During the Hospital Admission |
On the third day of admission, based on the clinical presentation and results above, the patient fulfilled the diagnostic criteria of HLH. Thus, more investigations were required to clarify whether it was primary or secondary, and the triggers (Table 2). In addition, although no CNS symptoms were present, a lumbar puncture was necessary. The lumbar puncture showed unremarkable cerebrospinal fluid (CSF) analysis (glucose, protein, culture, and cytology).
The management plan was to start HLH-94 protocol with dexamethasone, repeat the IVIG after 4 weeks, start Septra (trimethoprim/sulfamethoxazole) and nystatin mouthwash as prophylactic, cyclosporine and etoposide to be added if clinically needed. Moreover, we kept monitoring the clinical conditions and laboratory investigation with serial CBC, liver function test (LFT), fibrinogen, triglyceride, and ferritin levels.
Regarding the virology workup (Table 2), it showed positive rubella IgG, varicella IgG, CMV-IgG, HSV-IgM, HSV-IgG, COVID-19 IgG, and EBV antibodies. Thus, a consultation with the infectious disease team was done. After the discussion and based on EBV polymerase chain reaction (PCR) test, which came to be positive, they concluded that it was an acute EBV infection, and the positive serological testes for other viruses were considered a cross reactivity, and they advised to repeat the serological testes later.
After starting the patient on management protocol, the patient showed marvelous improvement. Clinically, he was afebrile and did not develop any new complaints. In addition, his labs were improving, and he was only maintained on dexamethasone without the need for other immunosuppressive or cytotoxic medications. During his hospital course, the patient did not develop any complications such as sepsis, bleeding, or others, and was uneventful. He was fit for discharge after 18 days of admission. Upon discharge, the examination was unremarkable. Labs showed CBC, renal function test (RFT), and LFT within the normal range, and ferritin level of 40.09. However, fibrinogen level was still 145, cholesterol level was 274 and triglyceride level was 250. He was discharged on dexamethasone and Bactrim (trimethoprim/sulfamethoxazole) with close follow-up in the clinic and IVIG sessions arranged.
Upon his follow-up visits, his labs were normalized (Table 3), and he continued on dexamethasone. In addition, the results of HLH genetic test panel by next generation sequencing showed a heterozygous mutation in STXBP2. So, he is a carrier of one of the familial HLH disease (type 5). However, as a carrier state, it cannot explain the clinical picture. Later, the result of primary immunodeficiency panel by next generation sequencing showed a hemizygous mutation in X-linked inhibitor of apoptosis protein (XIAP) along with another variant, which is consistent with the diagnosis of XLP-2. Thus, after reviewing the results, the mother was counseled regarding the bone marrow transplant (BMT), and he was referred to a transplant center. However, she was reluctant to do it, due to her not feeling convinced about the procedure and outcome, especially that he has no human leukocyte antigens (HLA)-matched related donor.
Click to view | Table 3. Trend of CBC, LFT, Triglycerides, Ferritin, and Fibrinogen Levels During Follow-Up Visits |
He had a follow-up for more than a year. Dexamethasone had been tapped off. The total duration of dexamethasone use from diagnosis until the last tapped dose is approximately 40 weeks. Between the follow-up visits, he was relapse-free, with a history of two admissions for gastroenteritis. However, it was not associated with major complications. Moreover, he was referred to the pediatric immunology clinic.
Our case shed light on the complexity of clinical practice. This patient turned out to be an XLP-2, STXBP2 carrier with extreme EBV infection and activation of HLH, who was successfully brought into remission by partial application of HLH-94 protocol and careful clinical and laboratory monitoring. The need for stem cell transplant (SCT) had been reinforced to the parents and he is still on follow-up with both.
| Discussion | ▴Top |
HLH, which is described as an excessive activation of the immune system, can be a life-threatening syndrome. Although significant advances were noted in understanding this disease, the diagnosis of HLH is still challenging due to nonspecific clinical presentation, overlapping with other inflammatory differential diagnoses or rapid deterioration of the patients due to multiple organ failure or death [6, 9]. As a result, these challenges resulted in delaying the diagnosis. A study conducted in 2021 showed that the median interval time between the onset of signs and symptoms of HLH and the diagnosis was 12 days, and the interval from the hospital admission until the diagnosis was 2 days [10]. In our case report, the diagnosis was made on the third day of admission, which is similar to the lateral study. This emphasizes the importance of listing HLH at the top of the differential list. Though it is a rare and complex disease, it can be fatal, and consideration of it as a differential will significantly impact the prognosis and outcome.
STXBP2 is one of the genetic mutations causing HLH, and it is part of familial hemophagocytic lymphohistiocytosis (FHL), precisely type 5 (FHL-5) [11]. However, our patient was only a carrier of this gene, with no expectation for it to cause any manifestations. On the other hand, XLP, with XLP-1 and XLP-2 as the most common subtypes, also causes HLH, and was detected in our patient. Regarding clinical presentation, XLP-2 has a clinical presentation and management approach is similar to the other genetic mutations causing HLH. However, it has a lesser severity and better prognosis [12-14]. Though EBV infection was the most common secondary cause of HLH [2], it is commonly associated with XLP-2 [12]. Despite the previous theories classifying HLH as primary and secondary, it has been noticed that many cases of primary HLH were triggered by secondary causes [6], and many cases of primary HLH are discovered in adulthood, especially by the presence of a secondary trigger [15]. This was also noted in our case, where our patient has both XLP-2 and heterozygous STXBP2 genetic mutations. However, EBV acted as the trigger for HLH to occur. The involvement of these two genes and having the HLH only to be triggered by EBV was not expected. Thus, changing this perspective may help widen the differential diagnosis spectrum and the clinical judgment. In addition, more studies are needed to further extend our knowledge about this syndrome.
COVID-19 infections have been linked to many diseases, including HLH. It has been hypothesized that COVID-19 infections can trigger the cytokine storm causing HLH [16]. There have been many reported cases of HLH triggered by COVID-19 infections. The characteristics of reported cases were variant; cases were reported in adults and pediatrics patients. HLH onset post COVID-19 occurred from days to months, and COVID-19 infection severity ranged from mild to severe [17-20]. All these findings suggest that COVID-19 can trigger HLH in different scenarios, and more studies are required to further understand the link between COVID-19 and HLH. Our patient had contact with COVID-19-confirmed case 1 week prior to the onset of HLH presentation, thus we cannot rule out the possibility of COVID-19, in addition to the other triggers, to cause HLH in our patient.
Generally, the goals of HLH management should focus on, firstly, stabilizing the patient. These patients can present with multi-organ failure or sepsis, which can be life-threatening. Thus, prompt management and supportive therapy are essential. Secondly, investigate the underlying cause of HLH, whether it is primary or secondary, and if secondary, search for the trigger, whether it is malignancy, infection, immune response or others. Thirdly, immune suppression is the core of HLH management [21]. In 1994, the Histiocyte Society commenced a trial (HLH-94), which was later used as a treatment protocol. The use of this protocol showed a dramatic improvement in survival. HLH-94 protocol includes the use of immunosuppressants (etoposide, corticosteroids, cyclosporin A, and, in specific patients, intrathecal methotrexate), which is essential to prevent further provoking of the immune system. Based on trials, physicians start with steroids, specifically dexamethasone, for better penetration into the CNS, in addition to a cytotoxic anti-cancer drug. The most used therapy includes dexamethasone with etoposide, which has been shown to be profoundly compelling and shows a better prognosis. In addition, the effects could be magnified when using cyclosporine in case of no response. Recently, etoposide replacement with antithymocyte globulin (ATG) has revealed notable improvement. Other drugs for relapsing HLH could be alemtuzumab, rituximab and interferon (IFN)-γ blocking monoclonal antibodies, which are under investigation [22-24]. In addition, gene therapy for the treatment of defective genes in HLH is under development, with the next step to be clinical trials [25]. Regarding our patient, he was managed using the HLH-94 protocol. However, he only required partial management. He has shown a significant improvement using only dexamethasone and supportive management, which was not expected while having two gene mutations and an acute EBV infection. Using this protocol, close monitoring and early identification of the disease helped in achieving a good outcome in our patient. Moreover, having the investigations going parallel with the management is timesaving and improves the outcome as seen in our patient.
SCT is the second step, after immunosuppressant of the HLH-94 protocol. SCT is considered a promising definitive therapy for most primary and secondary cases of HLH, which is regarded as a cure for relapsing cases too [21]. A study conducted in 2002 showed the 3-year probability of survival to be between 51% and 55% and to increase to 62% after SCT [23, 24]. However, recent studies showed there are several factors that decide the success of this cure, such as some genes have resulted in better prognosis than others, specifically: PRF1, UNC13D, STXBP2, STX11, RAB27A, LYST and SH2D1A. Secondly, the source of bone marrow is one factor. As studies have shown that matched unrelated donor transplants recorded 70% survival compared with matched sibling transplants. In addition, the response to the initial therapy and achieving the first remission may improve the outcome of SCT [26-28]. However various challenges pose some hazards to this operation. The first one is the state of disease. Committing a SCT while the disease is active increases mortality rate, due to numerous side effects. Another one is the complications, some of which are found before that treatment initiation, like CNS involvement, while others like pneumonia, infections, organ failure, hemorrhages, and graft versus host disease (GVHD) could lead to poor prognosis. Eventually, it is the conditioning regimen. Choosing the correct plan in the correct time ensures that GVHD risk is diminished, with no other complications occurring. The issue is that drugs used produce different effects on others, until several studies indicated that the usage of alemtuzumab, fludarabine and melphalan guarantee better results (75% survival). Thus, proper planning, monitoring and enhancing the patient’s conditions might reduce the risk of these complications [26, 27]. The genetic mutation in our patient is not from the genes known with its better prognosis. However, his initial response to the management was satisfactory, which may yield a good outcome. In addition, the fact that no related matched donor was found might open the path for possible transplant in the future with a matched unrelated donor and give the chance to observe the outcome of this new approach.
Raising awareness about the SCT and its role, especially in the community, is needed, as it will help ease all the ambiguity, answer all the questions of the patient’s families, and help them in decision-making regarding SCT. A study conducted in 2015 in India showed that awareness of stem cells in general was 25% in Delhi, and 18% in Bareilly [29]. Another recent study conducted in 2022 in Saudi Arabia showed that approximately 57% have significant knowledge about SCT in relation to sickle cell disease, and 42% were willing to donate [30]. As seen in our case, the mother was reluctant about the decision of SCT, and not having enough awareness about it, which was one of the reasons.
Conclusions
Diagnosing HLH can be challenging due to the rarity of the syndrome, the nonspecificity of the signs and symptoms, and the fast deterioration nature of it. In addition to that, many cases of primary HLH are triggered by a secondary cause, which is different from what was commonly known for HLH. As a result, delaying or missing the diagnosis can worsen the prognosis and delay the treatment. Raising awareness of this syndrome among healthcare providers will facilitate running the investigations while simultaneously supporting and managing the patients.
Acknowledgments
None to declare.
Financial Disclosure
This research received no external funding.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Patient consent was waived due to the lack of identifiable patient information.
Author Contributions
Diagnosing and managing the patient: MA. Original draft preparation: SA and NA. Reviewing, editing and proof-reading the final version of the manuscript: MA, SA, and NA. All authors have read and agreed to the published version of the manuscript.
Data Availability
The data used to support the findings of this study are included within the article.
| References | ▴Top |
- Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. 2018;13:27-49.
doi pubmed - Morimoto A, Nakazawa Y, Ishii E. Hemophagocytic lymphohistiocytosis: Pathogenesis, diagnosis, and management. Pediatr Int. 2016;58(9):817-825.
doi pubmed - Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27(136):519-525.
doi pubmed pmc - Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, Yamamoto K, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86(1):58-65.
doi pubmed - Meeths M, Horne A, Sabel M, Bryceson YT, Henter JI. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer. 2015;62(2):346-352.
doi pubmed - Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, Hines M, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66(11):e27929.
doi pubmed pmc - Brisse E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: differences and similarities. Br J Haematol. 2016;174(2):203-217.
doi pubmed - Kim YR, Kim DY. Current status of the diagnosis and treatment of hemophagocytic lymphohistiocytosis in adults. Blood Res. 2021;56(S1):S17-S25.
doi pubmed pmc - Creput C, Galicier L, Buyse S, Azoulay E. Understanding organ dysfunction in hemophagocytic lymphohistiocytosis. Intensive Care Med. 2008;34(7):1177-1187.
doi pubmed - Li X, Yan H, Xiao Z, Zhang X, Huang J, Xiang ST, Zheng M, et al. Diagnostic time lag of pediatric haemophagocytic lymphohistiocytosis and patient characteristics: a retrospective cohort study. Front Pediatr. 2021;9:692849.
doi pubmed pmc - Cetica V, Santoro A, Gilmour KC, Sieni E, Beutel K, Pende D, Marcenaro S, et al. STXBP2 mutations in children with familial haemophagocytic lymphohistiocytosis type 5. J Med Genet. 2010;47(9):595-600.
doi pubmed pmc - Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, Kanegane H, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood. 2011;117(5):1522-1529.
doi pubmed - Xu T, Zhao Q, Li W, Chen X, Xue X, Chen Z, Du X, et al. X-linked lymphoproliferative syndrome in mainland China: review of clinical, genetic, and immunological characteristic. Eur J Pediatr. 2020;179(2):327-338.
doi pubmed pmc - Nichols KE, Marsh RA. The X-linked lymphoproliferative syndromes. In: Sullivan KE, Stiehm ER (eds). Stiehm’s Immune Deficiencies. Academic Press, Amsterdam; 2014:475-495.
doi - Wang Y, Wang Z, Zhang J, Wei Q, Tang R, Qi J, Li L, et al. Genetic features of late onset primary hemophagocytic lymphohistiocytosis in adolescence or adulthood. PLoS One. 2014;9(9):e107386.
doi pubmed pmc - Opoka-Winiarska V, Grywalska E, Rolinski J. Could hemophagocytic lymphohistiocytosis be the core issue of severe COVID-19 cases? BMC Med. 2020;18(1):214.
doi pubmed pmc - Wiseman D, Lin J, Routy JP, Samoukovic G. Haemophagocytic lymphohistiocytosis in an adult with postacute COVID-19 syndrome. BMJ Case Rep. 2021;14(9):e245031.
doi pubmed pmc - Greenmyer JR, Wyatt KD, Milanovich S, Kohorst MA, Ferdjallah A. COVID-19-associated secondary hemophagocytic lymphohistiocytosis requiring hematopoietic cell transplant. EJHaem. 2022;3(3):1025-1028.
doi pubmed pmc - Mohamed Jiffry MZ, Ahmed-khan MA, Vargas JA, Thomas T, Josey S. Hemophagocytic lymphohystiocytosis in a patient with post-acute COVID-19 infection. Blood. 2022;140(Supplement 1):11183-11183.
doi - Molina G, Contreras R, Coombes K, Walgamage T, Perozo MA, DesBiens MT. Hemophagocytic lymphohistiocytosis following COVID-19 infection. Cureus. 2023;15(1):e34307.
doi pubmed pmc - Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, Gadner H, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373.
doi pubmed - Madkaikar M, Shabrish S, Desai M. Current updates on classification, diagnosis and treatment of hemophagocytic lymphohistiocytosis (HLH). Indian J Pediatr. 2016;83(5):434-443.
doi pubmed - George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med. 2014;5:69-86.
doi pubmed pmc - Carter-Febres M, Lozano-Chinga M, Thomsen W, Treemarcki EB, James KE, Fluchel M. Variation of diagnostic approaches and treatment practices for hemophagocytic lymphohistiocytosis/macrophage activation syndrome among pediatric subspecialists. J Pediatr. 2023;255:65-71.e66.
doi pubmed - Jordan M, Carmo M, Tiwari S, Arumugam P, Risma K, Gaspar B, Malik P. Gene therapy for hemophagocytic lymphohistiocytosis (HLH): Fixing a criticial “circuit breaker” in the immune system. Blood. 2012;120(21):3158-3158.
doi - Marsh RA, Jordan MB, Filipovich AH. Reduced-intensity conditioning haematopoietic cell transplantation for haemophagocytic lymphohistiocytosis: an important step forward. Br J Haematol. 2011;154(5):556-563.
doi pubmed pmc - Marsh RA, Haddad E. How i treat primary haemophagocytic lymphohistiocytosis. Br J Haematol. 2018;182(2):185-199.
doi pubmed - Bergsten E, Horne A, Hed Myrberg I, Arico M, Astigarraga I, Ishii E, Janka G, et al. Stem cell transplantation for children with hemophagocytic lymphohistiocytosis: results from the HLH-2004 study. Blood Adv. 2020;4(15):3754-3766.
doi pubmed pmc - Saran M, et al. Knowledge and awareness of stem cells among expectant mothers and parents of elementary school children in Bareilly and Delhi cities. Journal of Indian Association of Public Health Dentistry. 2015;13(4):502-508.
doi - Hurissi E, Hakami A, Homadi J, Kariri F, Abu-Jabir E, Alamer R, Mobarki R, et al. Awareness and acceptance of hematopoietic stem cell transplantation for sickle cell disease in Jazan Province, Saudi Arabia. Cureus. 2022;14(1):e21013.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.