

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 9, Number 2, June 2020, pages 50-54
Autoimmune Hepatitis With Severe Hypergammaglobulinemia and Eosinophilia in a Child
Shigeru Makinoa, Motokazu Nishikadoa, Hitoshi Awagunia, Ken-ichi Okumuraa, Jun Shinozukaa, Shinsaku Imashukua, b, c
aDivision of Pediatrics, Uji-Tokushukai Medical Center, Uji, Kyoto 611-0041, Japan
bDepartment of Laboratory Medicine, Uji-Tokushukai Medical Center, Uji, Kyoto 611-0041, Japan
cCorresponding Author: Shinsaku Imashuku, Division of Pediatrics, Uji-Tokushukai Medical Center, Uji, Kyoto 611-0041, Japan
Manuscript submitted May 2, 2020, accepted May 19, 2020, published online June 18, 2020
Short title: Autoimmune Hepatitis in a Child
doi: https://doi.org/10.14740/ijcp372
| Abstract | ▴Top |
We report here a 7-year-old Afghan girl with autoimmune hepatitis (AIH). Because she showed hepatic dysfunction with markedly high serum immunoglobulin G (IgG) (5,234 mg/dL) and eosinophilia (absolute eosinophil counts: 9,547/µL) at onset, we had difficulty in diagnosing this case by differentiating from various possible diseases. After wedge biopsy of the liver which histologically showed chronic hepatitis pattern as well as high titers (1:640) of anti-nuclear antibody with other negative autoantibodies, the patient was eventually diagnosed as AIH type 1 and successfully managed with a combination of prednisolone (PSL), azathioprine and ursodeoxycholic acid (UDCA), followed by a maintenance therapy with PSL, cyclosporine and UDCA.
Keywords: Autoimmune hepatitis; Hypergammaglobulinemia; Eosinophilia; Autoantibodies; Child
| Introduction | ▴Top |
Childhood autoimmune liver diseases include autoimmune hepatitis (AIH) and sclerosing cholangitis (ASC). Primary biliary cirrhosis (PBC), another hepatic autoimmune disease, is rare in children [1]. Generally, pediatric patients may present with either of the two autoimmune liver disorders, classical AIH or AIH/ASC overlap syndrome [2]. However, when the disease is associated with marked hypergammaglobulinemia and eosinophilia, the differential diagnosis of the disease is further difficult, because there are several possible diseases to be differentiated [3-6]. We report here a pediatric case of autoimmune hepatic disease, in which we encountered difficulties in reaching a definite diagnosis as well in the management of the disease.
| Case Report | ▴Top |
A 7-year-old Afghan girl living in Japan was hospitalized with headache and abdominal pain. Life in her family has been strictly in line with Islamic precepts. Prior to her admission, she had a complete (mandatory as well as optional) vaccination history and had no other remarkable previous medical history such as atopic dermatitis, asthma, or infectious events. No prescribed medicines were taken. Her parents were consanguineous (cousins) and she had three elder/one younger brothers and one elder sister. Mild-type β-thalassemia was diagnosed in her family members including her father, one of her elder brothers and elder sister. On admission, the patient, 115 cm in height and 20.4 kg in weight, was febrile (38 °C), with blood pressure 102/48 mm Hg, heart rate 124/min and respiration rate 22/min. Physically, she was noted to be anemic, with no heart murmur, no rales. Bilateral cervical lymph nodes were mildly enlarged while liver (1.5 cm) and spleen (2.0 cm) were palpable below the costal margin. The patient did not show any signs and symptoms of neurological abnormalities. Laboratory parameters were as follows: hemoglobin (Hb), 7.0 g/dL (reference: 11 - 16); mean corpuscular volume (MCV), 51.4 fL (83 - 100); mean corpuscular hemoglobin (MCH), 13.9 pg (28 - 36); mean corpuscular hemoglobin concentration (MCHC), 27.0% (31-36%); Mentzer index 10.2 (< 13.0 defined as indicating β-thalassemia than iron deficiency anemia); white blood cells (WBCs) 33,500/µL (3,000 - 8,500); and platelet count 508 × 103/µL (150 - 360 × 103/µL). Eosinophilia (28.5% of WBC; absolute eosinophil count, 9,547/µL) was noted. Liver enzymes were high including transaminases (aspartate aminotransferase (AST) 419 U/L (13 - 33) and alanine aminotransferase (ALT) 306 U/L (8 - 45)) as well as lactate dehydrogenase (LDH) and Gamma-glutamyl transpeptidase (γ-GTP), 293 U/L (122 - 228) and 320 U/L (8 - 33), respectively, while choline esterase (ChE) was low 53 U/L (206 - 477). The alkaline phosphatase/ALT ratio was 4.5. Other abnormal laboratory data included serum C-reactive protein (CRP) of 1.0 mg/dL (< 0.29), total bilirubin 2.17 mg/dL (0.3 - 1.3), total protein 9.6 g/dL (6.7 - 8.3), albumin 2.6 g/dL (4.1 - 5.2), immunoglobulin G (IgG) 5,234 mg/dL (820 - 1,740), IgA 437 mg/dL (90 - 100), and IgM 443 mg/dL (52 - 270) levels. Serum IgE was 557 U/mL (< 170), but none of IgE (radioallergosorbent test (RAST)) tests were positive. Renal function and coagulation data were normal. No active infectious disease was noted with negative hepatitis B surface antigen (HBsAg), hepatitis C virus antibody (HCVAb), parvovirus B19-Ab, human immunodeficiency virus (HIV)-Ab, and Epstein-Barr virus (EBV)-Ab. There was no parasite Abs. Abdominal computed tomography (CT) revealed hepato-splenomegaly, associated with remarkable periportal collar sign and edematous wall thickening of the gall bladder (Fig. 1). Based on significant polyclonal hypergammaglobulinemia with hepatic dysfunction, we first sought to differentiate between AIH [1] and idiopathic multicentric Castleman disease [3]. The serum interleukin-6 (IL-6) concentration was only 8 pg/mL (< 4). Tests for autoimmune Abs revealed the following: anti-nuclear Ab (antinuclear antibody (ANA); 1:640, homogeneous)-positive; anti-ds-DNA-Ab (-); anti-Sm-Ab (-); anti-mitochondria-Ab (-); anti-mitochondria M2-Ab (-); anti-smooth-muscle-Ab (SMA; -); anti-liver kidney microsome type 1 (anti-LKM1)-Ab (-); anti-liver cytosol type 1 (anti-LC1)-Ab (-); and soluble liver antigen/liver-pancreas (SLA/LP)-Ab (-). We also confirmed that she was negative in IgE (RAST) tests for any allergens including house dust, food, pollen and mites.
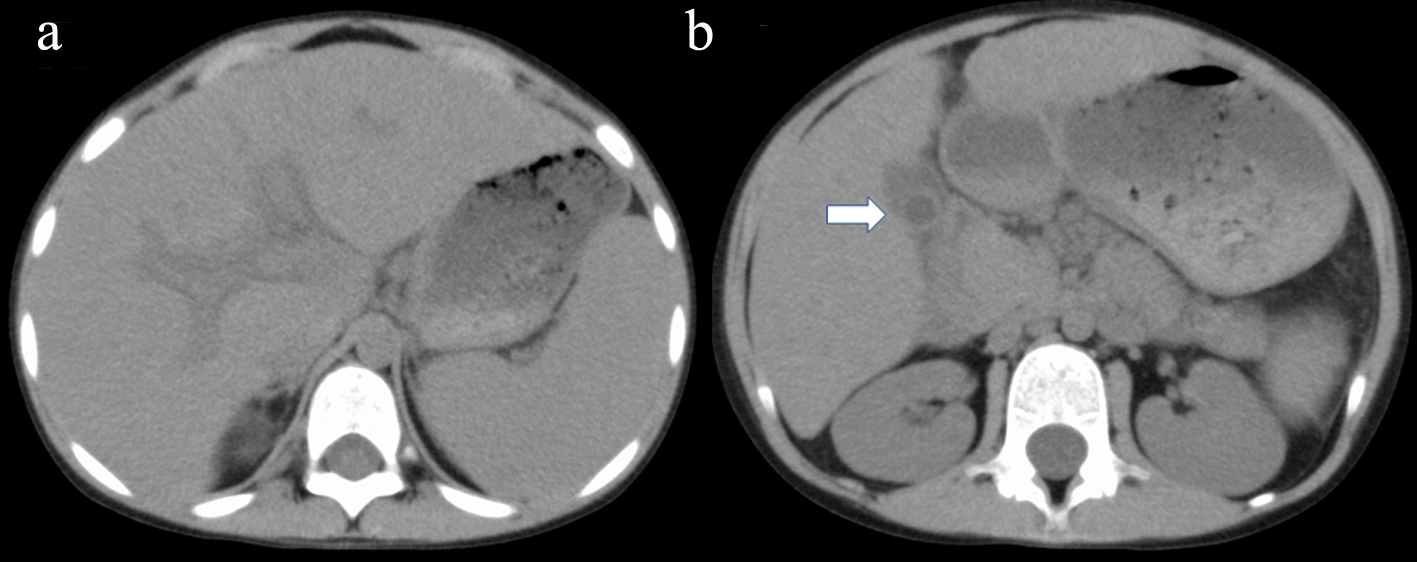 Click for large image | Figure 1. Abdominal axial CT shows hepato-splenomegaly associated with remarkable periportal collar sign (a) and edematous wall thickening of the gall bladder (b; arrow). CT: computed tomography. |
A laparoscopic wedge biopsy was obtained from the liver on day 11 post-admission; the histopathology revealed periportal CD3+ cells-dominant infiltration with aggregates of CD38+ or CD20+ cells in the portal area (Fig. 2). Eosinophils in the area were 2 - 3/hpf, Ki-67 in the lymphocytes was 5% and EBV-encoded RNA in situ hybridization (EBER-ISH) was negative. IgG4 was rarely stained (figure not shown). No findings indicating sclerosing cholangitis were present. As a result of histology, chronic hepatitis was suspected. In association with the significantly positive ANA results, we suspected AIH. Furthermore, diagnostic scores for AIH [7] as well as for AIH/ASC overlap syndrome [8] were calculated; scores following the original AIH scoring system were probable (13 points; as defined between 12 - 17 points) while those due to the simplified scoring system were definite (7 points; as defined ≥ 7 points) [7], while scores for the overlap syndrome were rejected (17 points; as defined < 19 points) [8]. Subsequently, this case was eventually diagnosed as AIH type 1, not AIH type 2 [2]. The patient was treated with oral ursodeoxycholic acid (UDCA; 50 mg × 3/day), followed by prednisolone (PSL; 0.6 mg/kg/day). After 4 weeks of treatment, serum transaminase levels fell markedly (to 20-30% of the initial values); however, the eosinophilia persisted. Therefore, the patient received azathioprine (AZP; 1.5 mg/kg/day, then tapered), UDCA and PSL (0.4 mg/kg/day, then tapered). One week after AZP, eosinophil counts were normalized. Serum IgG levels normalized by week 8 of treatment. Combined treatment with AZP/UDCA/PSL continued for 16 weeks. Thereafter, treatment was switched to oral cyclosporine (CSA; 2.5 mg/kg/day), UDCA, and PSL due to the relatively poor response of γ-GTP and ChE. At the 32 weeks of treatment, she is doing well, with normal serum IgG levels and no eosinophilia; however, liver function still remained abnormal (Fig. 3).
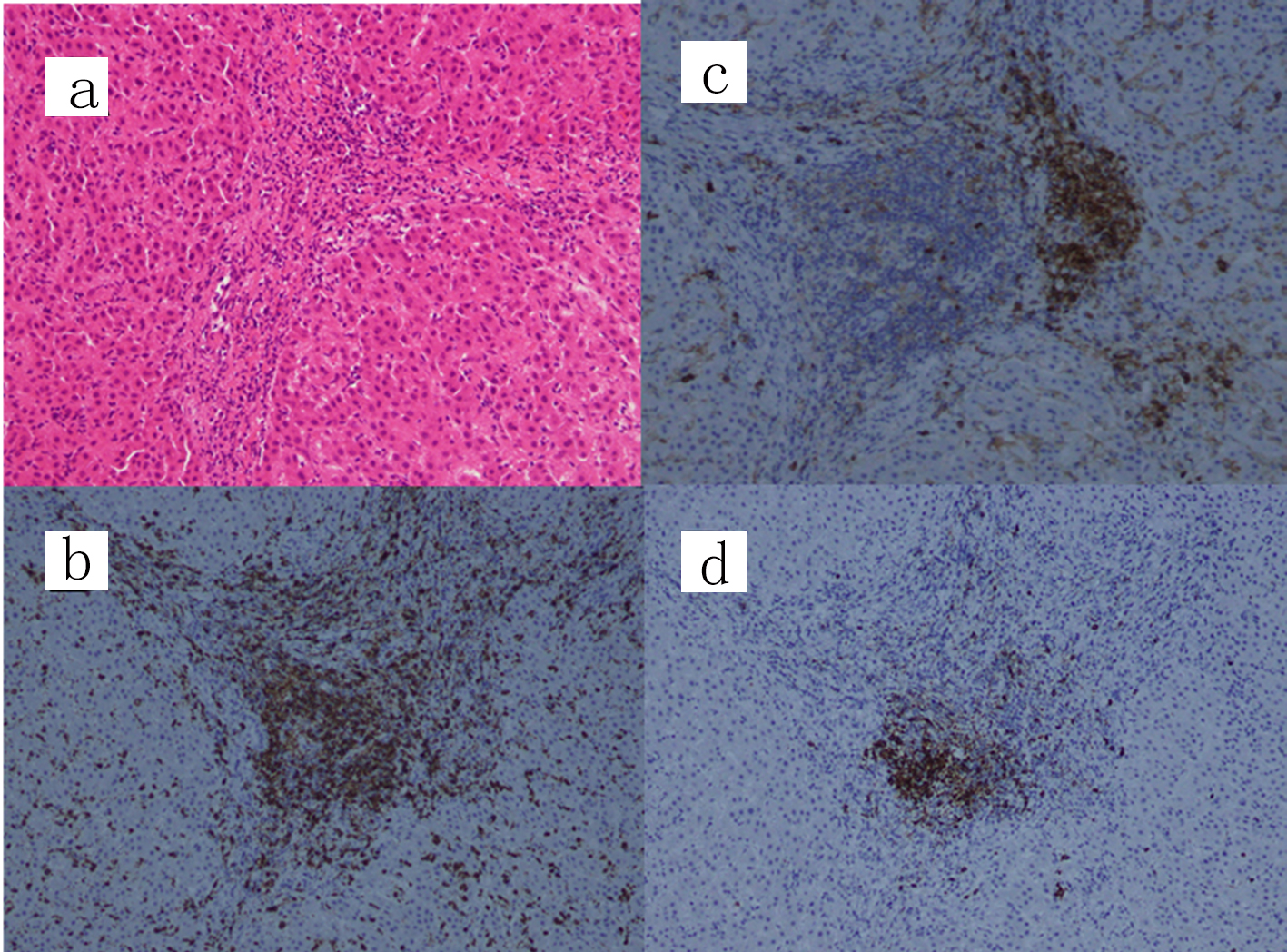 Click for large image | Figure 2. Liver wedge biopsy showing a chronic hepatitis pattern. (a) Hematoxylin and eosin staining, (b) CD3 staining, (c) CD38 staining and (d) CD20 staining (original magnification, × 100). IgG4 was scarcely stained and only a few eosinophils (2 - 3/hpf) are present at the portal area (figure not shown). IgG: immunoglobulin G. |
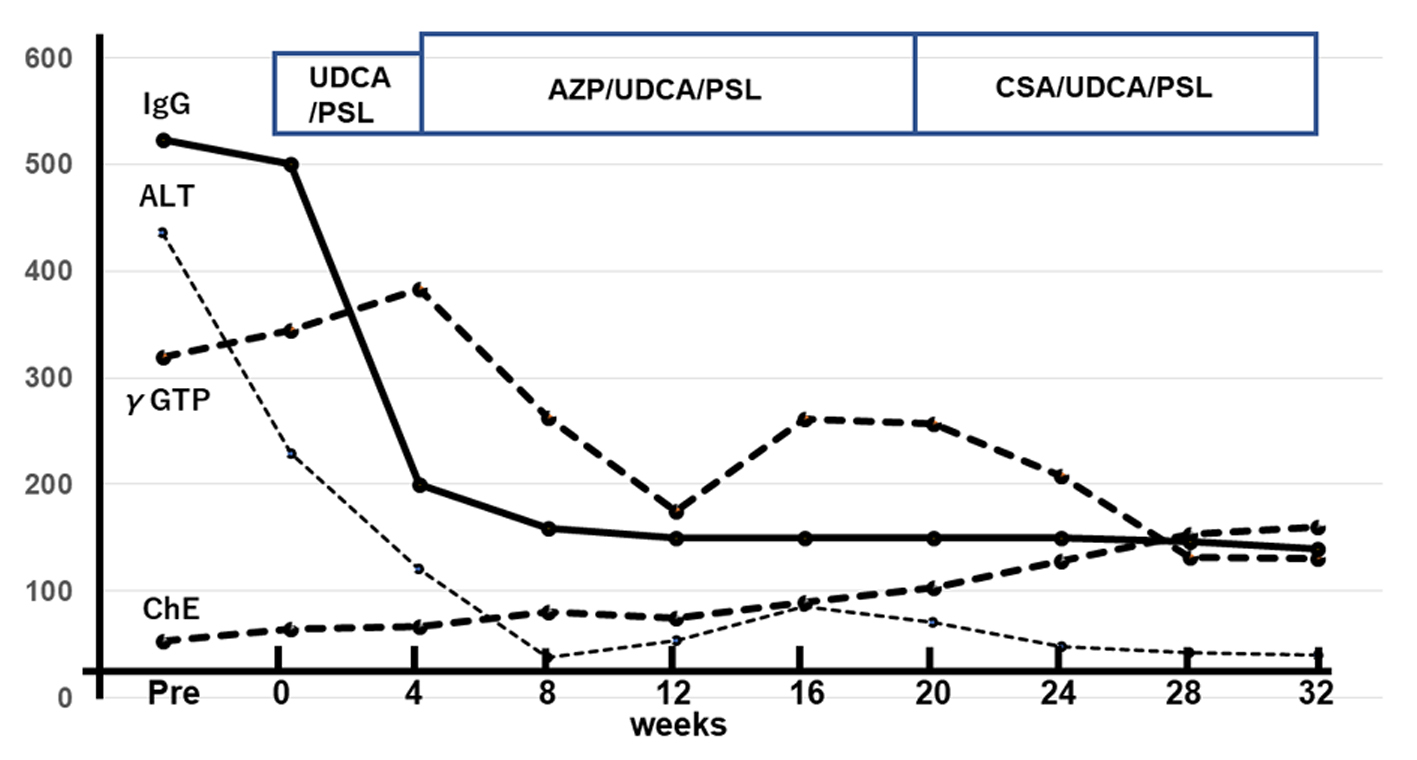 Click for large image | Figure 3. Time course of laboratory data showing improvement up to 32nd week of treatment. First, serum IgG and ALT levels normalized, followed by γ-GTP and ChE. Units on the vertical axis: IgG: × 10 mg/dL; ALT/γ-GTP/ChE, U/L. IgG: immunoglobulin G; ALT: alanine aminotransferase; γ-GTP: gamma-glutamyl transpeptidase; ChE: choline esterase. |
| Discussion | ▴Top |
This anemic 7-year old Afghan girl, with probable mild type β-thalassemia, exhibited hepatic dysfunction and eosinophilia with positive ANA associated with significantly elevated serum IgG. Her disease was thought to be acutely presented. Though we eventually reached a diagnosis of AIH type 1 by excluding various other possible diseases, we had difficulties to evaluate her hepatic disease, because AIH has diverse clinical phenotypes among ethnic groups [9]. As triggers of AIH, viral infections such as EBV were proposed [10]; however, in this case neither prior infectious events nor family history of autoimmunity were present.
In this case, liver histology revealed a chronic hepatitis pattern rather than an ASC-type pattern. In addition, ANA-positive/anti-SMA-negative autoantibody pattern suggested AIH type 1 and lack of anti-LKM1 and anti-LC1 Abs ruled out AIH type 2 [2]. In addition, the overlap syndrome [8] was also ruled out. An interesting finding in this case was the markedly high serum IgG level (> 5,000 mg/dL) and significant eosinophilia (> 9,000/µL) on admission. In such a condition, IgG4-related disease (IgG4-RD) needs to be differentiated [5, 6]. We did not assay serum IgG4 levels at onset, because pediatric IgG4-RD case is rare [11]; the diagnosis was later ruled out based on a scarce immunostaining of IgG4 in the biopsied liver tissue. In terms of significantly elevated serum IgG in our case, our literature survey on pediatric AIH cases revealed that most IgG values were in the ranges of 1,000 - 3,000 mg/dL or just said to be increased [12]. Serum IgG > 5,000 mg/dL in AIH seems to be rarely noted, but its significance related to the pathogenesis or outcome remains to be determined.
Another point of differential diagnosis was hypereosinophilia-related liver disease, since various types of liver injury including chronic active hepatitis were described in hypereosinophilic syndrome (HES) [4]. However, the diagnosis of HES could not be given because there was no previous history and eosinophils disappeared rapidly < 1.5 months after treatment. Considering the fact that she was negative for any allergens by IgE (RAST) tests as well as no history of asthma and atopic dermatitis, her eosinophilia might have been related to AIH, though in the biopsied liver tissue, eosinophils were not abundant. Thus, we ruled out the possibility like chronic hepatitis with eosinophilic infiltration associated with asthma, as previously reported [13]. In the context of autoimmune liver disease, eosinophilia can be a distinctive characteristic of early stage PBC [14], which has also been reported in cases of AIH [15]. A possible mechanism of eosinophilia in such cases may be due to mast cell activation; this can occur in cholestatic liver disease in which mast cell-derived mediators induce eosinophil chemotaxis and activation [14]. In our case, it was unknown why eosinophilia did not respond to PSL for 4 weeks’ treatment but quickly disappeared after 1 week of AZP. Precise cause of an association of eosinophilia with AIH remains to be clarified.
In the management, a combination of UDCA/PSL or of AZP/UDCA/PSL is the recommended treatment for AIH [16]. When AZP is prescribed, caution must be exercised for its bone marrow toxicity, thus the test of thiopurine methyltransferase (TPMT) activity before starting is preferred [17]. Our patient was treated initially with UDCA/PSL, which alone reduced serum IgG and ALT significantly; however, a combination treatment with AZP/UDCA/PSL was required to improve eosinophilia and ameliorate further liver dysfunction. In our case AZP was given safely without any toxicity, though we did not test TPMT activity.
Compared with adult AIH, pediatric AIH is said to be characterized by its aggressive course, but with appropriate immunosuppressive treatment 80% of patients can achieve remission. However, often due to nonadherence, relapses may occur; thus, long-term treatment is usually required, and only some 20% of AIH type 1 patients are able to discontinue therapy successfully [18]. As shown in Figure 3, our patient was on CSA/UDCA/PSL at the 32nd week of treatment. Thereafter, we have continued the same combination regimen and at the 47th week, her hepatic function nearly normalized except for γ-GTP. At this writing, it remains unknown how soon we can discontinue the treatment. In summary, in this pediatric case with rare AIH type 1 associated with serum IgG > 5,000 mg/dL and eosinophilia, we successfully managed with UDCA/PSL/AZP followed by UDCA/PSL/CSA after ruling out the overlap syndrome, HES- or IgG4-related disease.
Conclusions
Diagnosis and treatment of pediatric AIH are challenging. Liver histology and detailed autoantibody studies are prerequisites for diagnosis. Long-term careful management is required for preventing the progression into hepatic cirrhosis and failure, without disturbing the patient’s mental as well as physical growth by treatments.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
A written informed consent for publishing this case (including publication of images) was obtained from the parents of the patient.
Author Contributions
SM, MN, HA, KO and JS treated the patient; SM, MN and SI wrote the manuscript. All authors read and approved the final draft.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Maggiore G, Riva S, Sciveres M. Autoimmune diseases of the liver and biliary tract and overlap syndromes in childhood. Minerva Gastroenterol Dietol. 2009;55(1):53-70.
- Mieli-Vergani G, Vergani D. Autoimmune paediatric liver disease. World J Gastroenterol. 2008;14(21):3360-3367.
doi pubmed - Wang HW, Pittaluga S, Jaffe ES. Multicentric Castleman disease: where are we now? Semin Diagn Pathol. 2016;33(5):294-306.
doi pubmed - Minola E, Sonzogni A. Chronic hepatitis in hypereosinophilic syndrome: report of an unusual case. Infez Med. 2005;13(3):182-186.
- Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy. 2014;69(2):269-272.
doi pubmed - Culver EL, Sadler R, Bateman AC, Makuch M, Cargill T, Ferry B, Aalberse R, et al. Increases in IgE, eosinophils, and mast cells can be used in diagnosis and to predict relapse of IgG4-related disease. Clin Gastroenterol Hepatol. 2017;15(9):1444-1452 e1446.
doi pubmed - Sahebjam F, Vierling JM. Autoimmune hepatitis. Front Med. 2015;9(2):187-219.
doi pubmed - Zhang W, De D, Mohammed KA, Munigala S, Chen G, Lai JP, Bacon BR. New scoring classification for primary biliary cholangitis-autoimmune hepatitis overlap syndrome. Hepatol Commun. 2018;2(3):245-253.
doi pubmed - Czaja AJ. Autoimmune hepatitis in diverse ethnic populations and geographical regions. Expert Rev Gastroenterol Hepatol. 2013;7(4):365-385.
doi pubmed - Vento S, Cainelli F. Is there a role for viruses in triggering autoimmune hepatitis? Autoimmun Rev. 2004;3(1):61-69.
doi - Gillispie MC, Thomas RD, Hennon TR. Successful treatment of IgG-4 related sclerosing disease with rituximab: a novel case report. Clin Exp Rheumatol. 2015;33(4):549-550.
- Farid E, Isa HM, Al Nasef M, Mohamed R, Jamsheer H. Childhood autoimmune hepatitis in Bahrain: a tertiary center experience. Iran J Immunol. 2015;12(2):141-148.
- Omata F, Shibata M, Nakano M, Jacobs JL, Tokuda Y, Fukutake K, Takahashi O, et al. Chronic hepatitis with eosinophilic infiltration associated with asthma. Intern Med. 2009;48(22):1945-1949.
doi pubmed - Yamazaki K, Suzuki K, Nakamura A, Sato S, Lindor KD, Batts KP, Tarara JE, et al. Ursodeoxycholic acid inhibits eosinophil degranulation in patients with primary biliary cirrhosis. Hepatology. 1999;30(1):71-78.
doi pubmed - Chowdry S, Rubin E, Sass DA. Acute autoimmune hepatitis presenting with peripheral blood eosinophilia. Ann Hepatol. 2012;11(4):559-563.
doi - Liwinski T, Schramm C. Autoimmune hepatitis - update on clinical management in 2017. Clin Res Hepatol Gastroenterol. 2017;41(6):617-625.
doi pubmed - Lennard L. Implementation of TPMT testing. Br J Clin Pharmacol. 2014;77(4):704-714.
doi pubmed - Mieli-Vergani G, Vergani D. Autoimmune hepatitis in children: what is different from adult AIH? Semin Liver Dis. 2009;29(3):297-306.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.