

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 5, Number 3-4, December 2016, pages 51-53
Stars in Cytoplasm: Pediatric Neuroblastoma
Monazza Chaudharya, Shahzad Sarwara, Natasha Alia, b
aThe Aga Khan University Hospital, Karachi, Pakistan
bCorresponding Author: Natasha Ali, Department of Pathology and Laboratory Medicine/Oncology, The Aga Khan University Hospital, PO Box 3500, Stadium Road, Karachi 74800, Pakistan
Manuscript accepted for publication October 28, 2016
Short title: Pediatric Neuroblastoma
doi: https://doi.org/10.14740/ijcp260e
| Abstract | ▴Top |
We report a case of a 2-year-old female child who presented with abdominal distention and fever. On examination, there was a palpable mass in the right hypochondrium. Computed tomography of the abdomen revealed a large mass in the right suprarenal area. Biopsy of the mass revealed malignant round blue cell neoplasm. Bone marrow examination showed diffuse infiltration with atypical mononuclear cells and cytogenetic studies showed positivity for NMyc translocation.
Keywords: Neuroblastoma; NMyc; Abdominal distention
| Introduction | ▴Top |
Neuroblastoma (NB) is well known to be the most common malignant tumor in the pediatric age group under 1 year of age with around 650 cases every year in the United States [1]. It accounts for nearly 10% of all pediatric malignancies under the age of 15 and 15% mortality in that age group [2]. First described by Virchow in 1864 as “gliomas”, the disease has continued to bewilder physicians and scientists alike. The spectrum of its disease, in addition to its wide variety of manifestations, clinically enables it to be a clinical conundrum. The disease displays an erratic course that ranges from complete regression to the rapid development of a highly malignant tumor unresponsive to treatment. Due to advancements in molecular biology and genetic testing, certain characteristics and biomolecules have been identified; however, it has not changed the prognosis of the highly malignant form of the disease. Familial NB has been reported in 2% of cases where a mutation in the anaplastic lymphoma kinase (ALK) gene has been implicated [1]. The treatment of the disease requires a multidisciplinary approach including surgeons, oncologists, radiologists and bone marrow transplant specialists.
| Case Report | ▴Top |
A 2-year-old girl presented to the ER with a 1-week history of fever and abdominal pain, along with abdominal distension over the past few months. The fever was undocumented, continuous throughout the day, not relieved through antipyretics. It was associated with diffuse abdominal pain and reduced appetite. There were no associated symptoms of chills and rigors. Her past history was unremarkable, having had a normal birth with no complications. She was not on any medications and there was no history of family illnesses. On examination, the child appeared weak and pale. She was found to be tachycardic but afebrile. There was a palpable mass in the right hypochondrium along with significant tenderness. The rest of the systemic examination was unremarkable. Laboratory investigations revealed Hb 7 g/dl and Hct 25%, an elevated white count of 18 × 109/L, neutrophils 61% and lymphopenia. Platelet count was 242 × 109/L and LDH was elevated to 6,070 IU/L. Serum Ferritin was 1,125 ng/ml. Urine vanillylmandelic acid (VMA) was within normal range. LFTs, electrolytes and urine analysis were within normal limits. Computed tomography (CT) of the abdomen revealed a large mass in the right suprarenal area superiorly invading the liver parenchyma. Biopsy of the mass revealed malignant round blue cell neoplasm. Immunohistochemistry was positive for synaptophysin and CD56. Bone marrow examination showed diffuse infiltration with atypical mononuclear cells and cytogenetic studies showed positivity for NMyc translocation (Fig. 1).
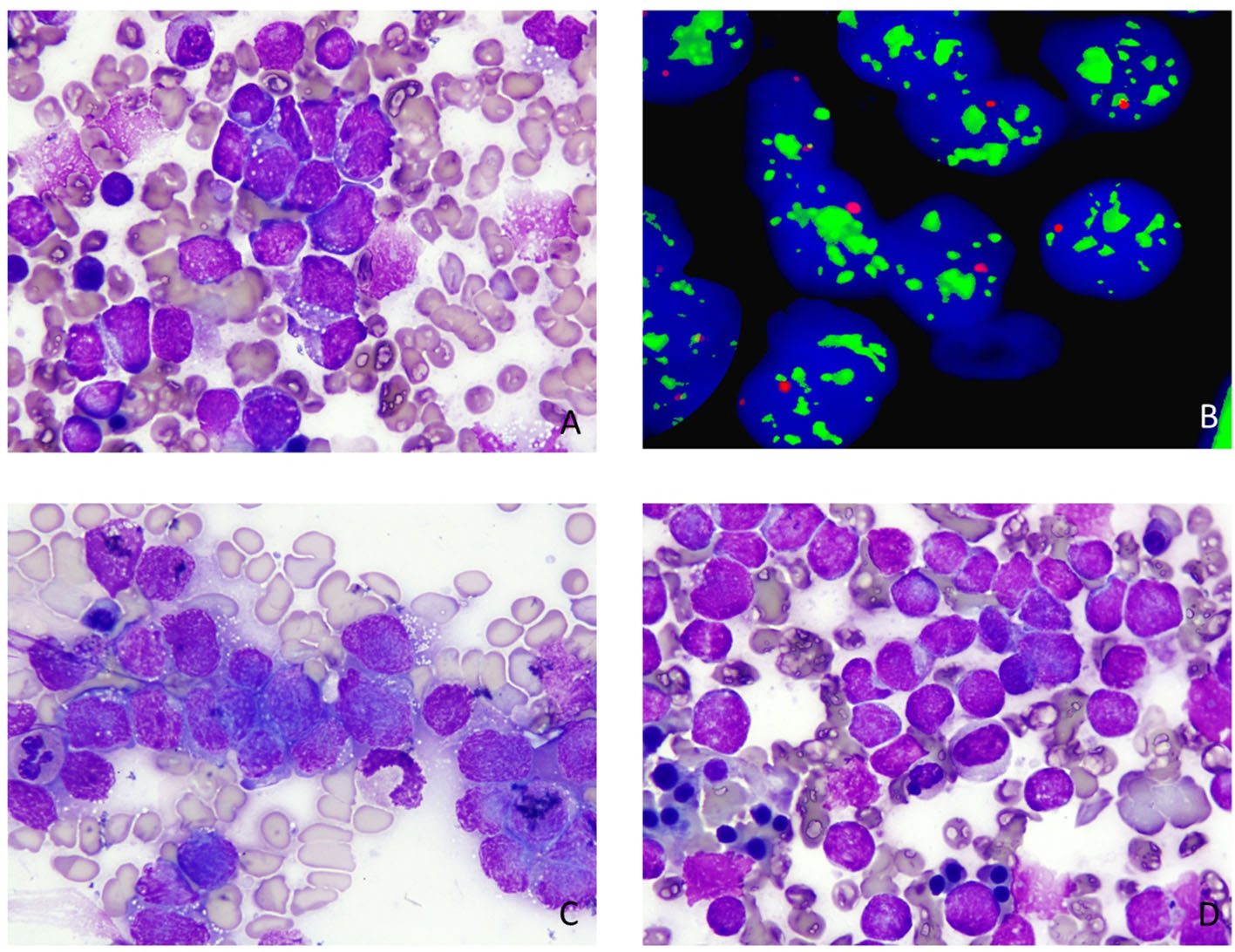 Click for large image | Figure 1. (A) Peripheral blood film (× 100) showing atypical mononuclear cells. (B) NMyc translocation by FISH. (C, D) Bone marrow specimen (× 100) showing atypical mononuclear cells with cytoplasmic vacuolations. |
| Discussion | ▴Top |
During early embryology, neural crest cells migrate to form the sympathetic tract extending from the base of the cranium to the pelvis forming the adrenal medulla, paraspinal sympathetic ganglia and sympathetic ganglia. A proliferation of these cells may occur anywhere in the region leading to a mass. The earliest description of NB is attributed to Virchow in 1864; however, it was in 1910 when James Homer Wright coined the term NB and described its characteristic histological feature that comprises of eosinophilic neuropils (neuritic processes) surrounded by neuroblasts, also known as Homer Wright rosettes. NB is part of a spectrum of ganglionic tumors characterized based on morphology that additionally include ganglioneuroblastoma and ganglioneuroma. In its early days, it was categorized into favorable or unfavorable based on its histology. The International Neuroblastoma Pathology Classification was devised that built on this earlier Shimada Classification to further aid in risk stratification in terms of prognosis.
Its clinical presentation is highly variant, as is the disease itself, depending on the location of the primary disease, any local invasion or metastatic involvement. Most commonly, it presents as an abdominal mass (75%), but it may also occur in the posterior mediastinum (20%) and pelvis (4%). There have been cases reported in unusual locations including the bladder and the mandible [3, 4]. The child may present with unexplained hypertension, diarrhea and weight loss as a result of the excess excretion of catecholamines such as VMA and vasoactive intestinal peptide (VIP). Ptosis, anhydrosis, and miosis (Horner’s syndrome) may develop due to compression from a cervical tumor. Respiratory difficulties may ensue from a thoracic mass. Metastasis is most commonly seen in the bone and bone marrow. Older children who develop metastatic disease may present with bone pain or periorbital ecchymoses (or raccoon eyes) depicting infiltration into the retroorbital venous plexus. Left ventricular metastasis has also been reported [5]. Dancing eye syndrome or opsoclonus myoclonus syndrome, a paraneoplastic syndrome that involves nystagmus with multidirectional eye movements, myoclonus and ataxia, was seen to have a 20% occurrence in NB [6]. It serves as a good prognostic indicator in terms of progression to malignancy; however, children with this manifestation were seen to have long-term learning disabilities and developmental delays with relapsing of symptoms.
Laboratory tests include liver and kidney function tests, urinary or serum catecholamine metabolites including VMA, and homovanillic acid (HVA) which are elevated in 90% cases of NB. Lactate dehydrogenase (LDH) and ferritin may also be elevated and are used to assess treatment. Owing to the incidence of the disease and its ominous nature and detectability in urinary metabolites, many countries including Japan, Germany, Denmark and North America ran mass screening tests over 20 years at different pediatric age groups including 3 weeks of age and then at 6 months. However, this showed no change in terms of prognosis of the disease as there were some cases that involved spontaneous regression that would have not required any treatment to begin with [7]. Both CT and magnetic resonance imaging (MRI) remain the modalities of choice with the latter being superior in terms of sensitivity (83% vs. 43% in CT), as well as in the detection of metastasis and intraspinal extension. When evaluating involvement of bone and bone marrow, scintigraphic imaging using metaiodobenzylguanidine (MIBG) has replaced earlier modalities but may not be available in every institution [1].
Tissue biopsy is assessed in accordance with the International Neuroblastoma Pathology Classification (INPC) in terms of cellular turnover (mitosis-karyorrhexis index (MKI)), presence of Schwannian stroma and the differentiation of neuroblasts. It is classified according to age and thus aids in the prognosis of the disease [1].
A definitive diagnosis of NB is obtained either through histological diagnosis of the tissue by light microscopy with or without immunohistochemical assay, electron microscopy, or elevated urinary catecholamines or through metastasis-proven bone marrow aspirate with elevated urinary or serum catecholamine metabolites.
Advancements in molecular biology have aided in terms of prognosis and understanding of the heterogenous nature of the disease. Amplification of the MYCN has been strongly implicated in the development of NB and is used as a prognostic indicator to determine the sporadic, aggressive tumors in its advanced stages. The anaplastic lymphoma kinase (ALK) gene has also been implicated in familial NB cases occurring in around half [1].
The International Neuroblastoma Risk Group Staging System (INRSS) is the most widely used classification system and uses biochemical, tissue diagnosis and imaging to categorize and stage the disease, thereby allowing appropriate management in accordance with the stage of the disease [8]. The disease is stratified according to risk into low, intermediate and high, with low risk showing favorable outcomes of event free survival rates as high as 95%, whereas the high risk group shows poor outcomes of less than 50%. Treatment ranges from local resection to chemotherapy agents including cyclophosphamide, doxorubicin, as well as radiation therapy.
Although decades have passed since its first discovery, the prognosis of high risk NB remains unchanged [9]. Molecular biochemical and genetic research shows a promising avenue for future treatment in the management of NB and may shed light on its pathogenesis and erratic nature.
Conflicts of Interest
The authors declare no conflicts of interest.
Author Contributions
MC drafted manuscript, SS took pictures and drafted manuscript, and NA conceived idea and critically reviewed manuscript.
| References | ▴Top |
- Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930-935.
doi pubmed - Colombani PM, Foglia RP, Skinner M A. Principles and practice of pediatric surgery (Vol. 1504). K. T. Oldham (Ed.). Lippincott Williams & Wilkins. 2005.
- Kojima S, Yagi M, Asagiri K, Fukahori S, Tanaka Y, Ishii S, Saikusa N, et al. Infantile neuroblastoma of the urinary bladder detected by hematuria. Pediatr Surg Int. 2013;29(7):753-757.
doi pubmed - Tang PH, Cohen PA. Primary neuroblastoma of the mandible. Singapore Med J. 2009;50(1):e5-7.
pubmed - Gupta D, Srikanthan M, Lagmay J, Co-Vu JG. Left Ventricular Metastasis in Neuroblastoma: A Case Report. J Pediatr Hematol Oncol. 2016;38(1):74-77.
doi pubmed - Brunklaus A, Pohl K, Zuberi SM, de Sousa C. Outcome and prognostic features in opsoclonus-myoclonus syndrome from infancy to adult life. Pediatrics. 2011;128(2):e388-394.
doi pubmed - Schilling FH, Spix C, Berthold F, Erttmann R, Fehse N, Hero B, Klein G, et al. Neuroblastoma screening at one year of age. N Engl J Med. 2002;346(14):1047-1053.
doi pubmed - Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. 2009;27(2):298-303.
doi pubmed - Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clin Cancer Res. 2012;18(10):2740-2753.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.