

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 4, Number 2-3, October 2015, pages 154-157
Congenital Hyperinsulinism and Cochlear Hypoplasia in a Rare Case of Pallister-Hall Syndrome
Dinesh Giria, Shivaram Avulab, Victoria McKayc, Astrid Weberc, Mo Didia, Senthil Senniappana, d
aDepartment of Paediatric Endocrinology, Alder Hey Children’s Hospital NHS Trust, Eaton Road, Liverpool, L12 2AP, UK
bDepartment of Paediatric Radiology, Alder Hey Children’s Hospital NHS Trust, Eaton Road, Liverpool, L12 2AP, UK
cDepartment of Clinical Genetics, Liverpool Women’s Hospital, Crown Street Liverpool, L8 7SS, UK
dCorresponding Author: Senthil Senniappan, Department of Paediatric Endocrinology, Alder Hey Children’s Hospital NHS Trust, Liverpool, UK
Manuscript accepted for publication March 20, 2015
Short title: Hyperinsulinemic Hypoglycemia
doi: http://dx.doi.org/10.14740/ijcp196w
| Abstract | ▴Top |
Pallister-Hall syndrome (PHS) is characterized by a spectrum of anomalies, which include polydactyly, hypothalamic hamartoma, laryngotracheal cleft, bifid epiglottis, imperforate anus, and renal abnormalities. Hypoplastic cochlea is a rare reported association of PHS. A baby girl was born at 31 weeks gestation with birth weight of 1.2 kg. The endotracheal intubation was extremely difficult due to narrow trachea. She was noted to be dysmorphic and have recurrent hypoglycemic episodes requiring high concentration of glucose infusion. The investigations revealed an inappropriately high plasma insulin and C-peptide level during hypoglycemia with low free fatty acids and β-hydroxy butyrate suggestive of congenital hyperinsulinism (CHI). MRI of the brain revealed the presence of a large hypothalamic hamartoma. The cochlea was noted to be truncated bilaterally with reduced number of turns. The cytogenetic analysis revealed GLI3 mutation consistent with diagnosis of PHS. This is the first reported case of CHI in association with PHS. Our patient also had hypoplastic cochlea, which is a unique but rarely reported feature of PHS.
Keywords: Congenital hyperinsulinism; Pallister-Hall syndrome; Cochlear hypoplasia
| Introduction | ▴Top |
Congenital hyperinsulinism (CHI) is the most common cause of persistent and recurrent hypoglycemia in infancy [1]. It is the result of unregulated insulin secretion from the pancreatic β cells leading to severe hypoglycemia [2]. CHI is caused by genetic defects in key genes regulating insulin secretion. The genetic basis of CHI involves mutations in nine different genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, HNF4A, HNF1A and UCP2), which regulate insulin secretion from the pancreatic β cells [2, 3]. It can also be secondary to risk factors like birth asphyxia, intra-uterine growth retardation, Rh isoimmunisation and maternal diabetes mellitus or associated with various developmental syndromes [2, 3].
Pallister-Hall syndrome (PHS) is characterized by a spectrum of anomalies ranging from polydactyly, hypothalamic hamartoma, laryngotracheal cleft, bifid epiglottis, imperforate anus, and renal abnormalities [4]. This syndrome is caused by the mutation of GLI3 and is inherited in an autosomal dominant fashion [4]. We report a rare case of CHI in PHS. The association of PHS with CHI has not been previously reported in the literature. The patient was also found to have hypoplastic cochlea, which has been reported only once in association with PHS [5].
| Case Report | ▴Top |
A baby girl was born by elective section at 31 weeks of gestation with a birth weight of 1.2 kg (-1.76 SDS) following concerns regarding intra-uterine growth retardation and oligohydramnios. She was the first child to non-consanguineous Caucasian parents. The baby was born in a reasonable condition (APGAR scores of 5 and 8 at 1 and 10 min respectively). Soon after the delivery, baby was noted to be pale with irregular respirations and desaturations. Endotracheal intubation was extremely difficult with multiple unsuccessful attempts as the trachea was described to be very narrow during attempted intubation. The baby was stabilized on continuous positive airway pressure (CPAP) and transferred to pediatric intensive care. She was then intubated with difficulty with a size 2.0 endotracheal tube. The examination of the upper airway showed a bifid epiglottis, uvula and a funnel shaped trachea.
The baby had oligodactyly with three fingers and a thumb on the right hand and four fingers and a thumb on the left hand. She had short fingers and a single palmar crease bilaterally. Toes were noted to be overlapping with slightly rocker-bottom appearances to the feet. She also had a short nose with a depressed nasal bridge and anteverted nares. She had two umbilical vessels, an imperforate anus and a small urogenital opening. An ileostomy pouch was created for the imperforate anus. A cardiac echocardiogram revealed a small patent ductus arteriosus but otherwise a structurally normal heart. High resolution CT scan of the chest revealed a trachea of very small diameter of 3.5 mm. A bronchogram demonstrated the right upper lobe bronchus arising from the trachea and small airways. MRI scan of the brain revealed the presence of a large mass in the suprasellar space measuring 25 × 17 × 22 mm in size with imaging characteristics of a hypothalamic hamartoma (Fig. 1). The cochlea was noted to be truncated bilaterally with reduced number of turns (Fig. 2). The intensive care stay was further complicated with the baby developing pneumothorax and requiring a chest drain.
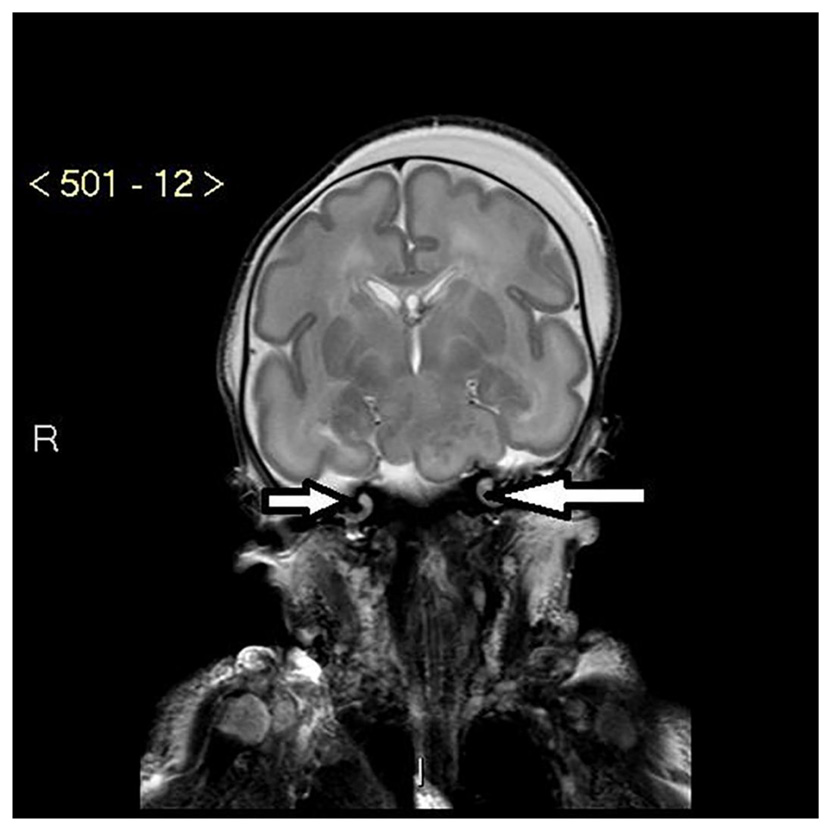 Click for large image | Figure 1. MRI of bilateral hypoplastic cochlea with reduced number of apical and basal turns (white arrows). |
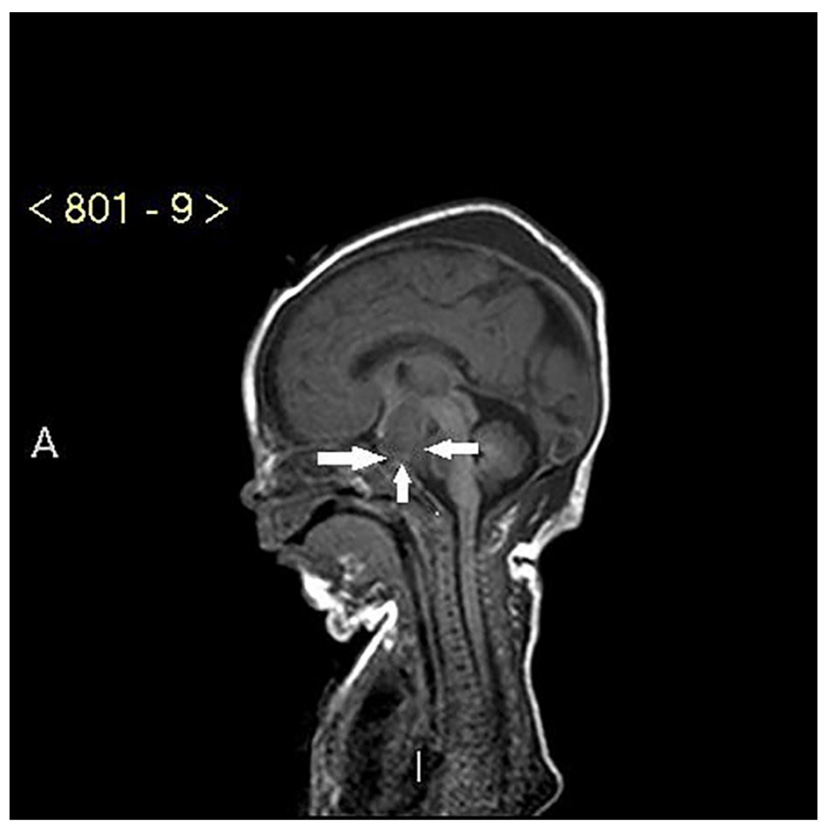 Click for large image | Figure 2. MRI of large hypothalamic hamartoma (white arrows). |
The baby was found to have recurrent hypoglycemia (blood glucose < 2.6 mmol/L) from the first day of life requiring high concentration glucose infusion to maintain blood glucose greater than 3.5 mmol/L (glucose infusion rate 18 mg/kg/min). The investigations undertaken to investigate hypoglycemia showed inappropriately elevated plasma insulin level of 65 pmol/L and C-peptide of 775 pmol/L when the blood glucose level was 1.4 mmol/L. The plasma free fatty acids (285 µmol/L) and 3-hydroxy butyrate (8 µmol/L) were suppressed during hypoglycemia, all features confirming a diagnosis of CHI.
The baby was subsequently commenced on diazoxide at a dose of 5 mg/kg/day along with chlorothiazide at 7 mg/kg/day. The baby’s glucose requirement gradually reduced and normoglycemia (blood glucose > 3.5 mmol/L) was subsequently maintained on enteral feeds. The baby was found to tolerate diazoxide well without side effects such as fluid retention.
A quantitative fluorescence-polymerase chain reaction (QF-PCR) analysis was performed for trisomies 21, 18 and 13 and was negative. The microarray analysis did not identify copy number variants. The phenotypic features of syndactyly, airway malformation including bifid epiglottis, imperforate anus, solitary kidney and the MRI evidence of hypothalamic hamartoma and hypoplastic cochlea pointed towards a diagnosis of PHS. The sequencing of GLI3 revealed a pathogenic G to T nucleotide substitution in exon 14 of GLI3, c.3439G>T, which confirmed the diagnosis of PHS. The mutation was subsequently confirmed to have arisen de novo.
| Discussion | ▴Top |
PHS (OMIM 146510) is a pleiotropic disorder of human development that comprises hypothalamic hamartoma, central polydactyly and other malformations (Table 1) [4]. It was first described in 1980 in six newborn babies [6, 7]. Hypothalamic hamartoma is often asymptomatic; however, it may be associated with pan hypopituitarism [8]. Renal agenesis or dysplasia as well as other genitourinary anomalies has been reported, including vaginal atresia, hydrometrocolpos, microphallus, and cryptorchidism [9]. Neurological involvement can include epilepsy. Precocious puberty has also been reported in patients with PHS [10]. Generalized skeletal dysplasia with mesomelic shortening and radial bowing of limbs has been reported [11]. The clinical features of PHS have been summarized in Table 1.
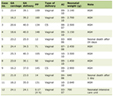 Click to view | Table 1. Clinical Features of Pallister-Hall Syndrome [4] |
PHS is inherited in an autosomal dominant manner [6]. About 25% of cases are caused by de novo mutations and the clinical presentation is generally more severe than the familial cases [4]. PHS is caused by mutations in GLI3, which encodes a 1590 amino acid zinc-finger transcription factor on chromosome 7p14.1 [12]. It is an essential component of the Sonic Hedgehog (SHH) signaling pathway in mammalian skeletogenesis [13]. GLI3 has a dual function as a transcriptional activator and a repressor of the SHH pathway, and plays a role in limb development [12, 13]. The other autosomal dominant skeletal dysplasia associated with GLI3 mutation is Graig cephalopolysyndactyly syndrome (GCPS) (OMIM 175700), caused by loss-of-function mutations spread across the gene [12, 13]. This syndrome is characterized by preaxial polydactyly, macrocephaly and other features including postaxial polydactyly, syndactyly, and hypertelorism [12, 13].
Whilst abnormal cochleae have been identified in mouse models of PHS [14], cochlear abnormalities in humans have been described in the literature rarely. We identified one case report describing a case of PHS in a girl with bilateral cochlear hypoplasia on MRI [5]. GLI3 mutation can cause cochlear malformations as the GLI3 and SHH pathways play an important role in the development of the cochlea [15]. Though the MRI scan was aimed at evaluating the brain, cochlear hypoplasia was clearly demonstrated in our case. We believe that a hypoplastic cochlea could represent a surrogate marker of PHS in individuals without hypothalamic hamartomas or typical multiple congenital anomalies and dedicated MRI of the cochlea can contribute to the diagnosis of these children.
Our patient also had CHI, which has been described in the literature in association with various syndromes (Table 2) [3] such as Beckweth-Wiedemann syndrome [16], Soto’s syndrome [17], Usher’s syndrome [18], and Costello syndromes [3]. However, the association of CHI in PHS has not been previously reported in the literature. GLI3 is implicated in the cochlear abnormality and the digital abnormalities described in PHS. We have not been able to establish the exact genetic link between GLI3 mutation and CHI, but SHH pathways that are modified by GLI3 have been implicated in the early development of the pancreas as well as subsequent insulin activity in pancreatic β cells [19, 20]. Further studies are needed to establish the exact genetic link between GLI3 mutation and CHI.
Click to view | Table 2. Syndromes Associated with Congenital Hyperinsulinism [3] |
Conclusion
PHS is a rare genetic disorder with many systemic abnormalities reported in the literature. This is the first reported case of CHI in association with this syndrome. The patient also had the typical cochlear abnormality in PHS, which can be used as a surrogate feature in the diagnosis of PHS.
Conflict of Interest
None.
| References | ▴Top |
- Soltesz GA. Hyperinsulinism in infancy and childhood. Ergeb Inn Med Kinderheilkd. 1984;51:115-202.
doi - Senniappan S, Arya VB, Hussain K. The molecular mechanisms, diagnosis and management of congenital hyperinsulinism. Indian J Endocrinol Metab. 2013;17(1):19-30.
doi pubmed - Mohamed Z, Arya VB, Hussain K. Hyperinsulinaemic hypoglycaemia:genetic mechanisms, diagnosis and management. J Clin Res Pediatr Endocrinol. 2012;4(4):169-181.
doi pubmed - Biesecker LG, Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. Source Gene Reviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014.
- Avula S, Alam N, Roberts E. Cochlear abnormality in a case of Pallister-Hall syndrome. Pediatr Radiol. 2012;42(12):1502-1505.
doi pubmed - Hall JG, Pallister PD, Clarren SK, Beckwith JB, Wiglesworth FW, Fraser FC, Cho S, et al. Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus and postaxial polydactyly--a new syndrome? Part I: clinical, causal, and pathogenetic considerations. Am J Med Genet. 1980;7(1):47-74.
doi pubmed - Clarren SK, Alvord EC, Jr., Hall JG. Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly--a new syndrome? Part II: Neuropathological considerations. Am J Med Genet. 1980;7(1):75-83.
doi pubmed - Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124(1):113-123.
pubmed - Narumi Y, Kosho T, Tsuruta G, Shiohara M, Shimazaki E, Mori T, Shimizu A, et al. Genital abnormalities in Pallister-Hall syndrome: Report of two patients and review of the literature. Am J Med Genet A. 2010;152A(12):3143-3147.
doi pubmed - Boudreau EA, Liow K, Frattali CM, Wiggs E, Turner JT, Feuillan P, Sato S, et al. Hypothalamic hamartomas and seizures: distinct natural history of isolated and Pallister-Hall syndrome cases. Epilepsia. 2005;46(1):42-47.
doi pubmed - Roscioli T, Kennedy D, Cui J, Fonseca B, Watson GF, Pereira J, Xie YG, et al. Pallister-Hall syndrome: unreported skeletal features of a GLI3 mutation. Am J Med Genet A. 2005;136A(4):390-394.
doi pubmed - Johnston JJ, Olivos-Glander I, Killoran C, Elson E, Turner JT, Peters KF, Abbott MH, et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am J Hum Genet. 2005;76(4):609-622.
doi pubmed - Biesecker LG. What you can learn from one gene: GLI3. J Med Genet. 2006;43(6):465-469.
doi pubmed - Driver EC, Pryor SP, Hill P, Turner J, Ruther U, Biesecker LG, Griffith AJ, et al. Hedgehog signaling regulates sensory cell formation and auditory function in mice and humans. J Neurosci. 2008;28(29):7350-7358.
doi pubmed - Bok J, Dolson DK, Hill P, Ruther U, Epstein DJ, Wu DK. Opposing gradients of Gli repressor and activators mediate Shh signaling along the dorsoventral axis of the inner ear. Development. 2007;134(9):1713-1722.
doi pubmed - Adachi H, Takahashi I, Higashimoto K, Tsuchida S, Noguchi A, Tamura H, Arai H, et al. Congenital hyperinsulinism in an infant with paternal uniparental disomy on chromosome 11p15: few clinical features suggestive of Beckwith-Wiedemann syndrome. Endocr J. 2013;60(4):403-408.
doi pubmed - Matsuo T, Ihara K, Ochiai M, Kinjo T, Yoshikawa Y, Kojima-Ishii K, Noda M, et al. Hyperinsulinemic hypoglycemia of infancy in Sotos syndrome. Am J Med Genet A. 2013;161A(1):34-37.
doi pubmed - Al Mutair AN, Brusgaard K, Bin-Abbas B, Hussain K, Felimban N, Al Shaikh A, Christesen HT. Heterogeneity in phenotype of usher-congenital hyperinsulinism syndrome: hearing loss, retinitis pigmentosa, and hyperinsulinemic hypoglycemia ranging from severe to mild with conversion to diabetes. Diabetes Care. 2013;36(3):557-561.
doi pubmed - Thomas MK, Rastalsky N, Lee JH, Habener JF. Hedgehog signaling regulation of insulin production by pancreatic beta-cells. Diabetes. 2000;49(12):2039-2047.
doi pubmed - Mfopou JK, Baeyens L, Bouwens L. Hedgehog signals inhibit postnatal beta cell neogenesis from adult rat exocrine pancreas in vitro. Diabetologia. 2012;55(4):1024-1034.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.