

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 3, Number 4, December 2014, pages 117-120
Report of the First Moroccan Case of Fusion of Metacarpals 4/5 and Review of the Literature
Siham Chafai Elalaouia, b, d, Wafae Jdiouib, Soukaina Guaouaa, Imane Cherkaoui Jaouada, b, Tarik Madhic, Abdelaziz Sefiania, b
aCentre de Genomique Humaine, Faculte de Medecine et de Pharmacie, Universite Mohammed V Souissi, Rabat, Morocco
bDepartement de Genetique Medicale, Institut National d’Hygiene, Rabat, Morocco
cService de Chirurgie Orthopedique, Hopital d’Enfants, Rabat, Morocco
dCorresponding Author: Siham Chafai Elalaoui, Departement de Genetique Medicale, Institut National d’Hygiene, 27 Avenue Ibn Batouta, B.P. 769, 11400 Rabat, Morocco
Manuscript accepted for publication November 21, 2014
Short title: First Fusion of Metacarpals 4/5
doi: http://dx.doi.org/10.14740/ijcp176e
| Abstract | ▴Top |
Metacarpal 4-5 fusion (MF4; MIM #309630) is a very rare congenital malformation of the hand. It consists of a partial or complete fusion of the fourth and fifth metacarpals. This anomaly could be isolated or could be part of a genetic syndrome. Recently, FGF16 nonsense mutations were reported as the cause of isolated X-linked recessive MF4. We report an 18-month-old Moroccan boy with metacarpal 4-5 fusion for whom Sanger sequencing of the entire coding sequence of FGF16 was performed. FGF16 sequencing identified the already reported (c.C535T; p.R179X) mutation in exon 3 of the gene. The mutation was found in heterozygosity state in the clinically unaffected mother of the proband. Our finding showed that truncating mutation (c.C535T; p.R179X) of FGF16 causing X-linked recessive metacarpal 4-5 fusion is recurrent in patients from different ethnicity.
Keywords: FGF16; Metacarpal 4-5 fusion; X-linked inheritance; Recurrent mutation
| Introduction | ▴Top |
Metacarpal 4-5 fusion (MF4; MIM # 309630) is a rare congenital bone malformation caused by mutation in the FGF16 gene on chromosome Xq21. Phenotypically, it is characterized by ulnar deviation of both fifth fingers, bilateral clinodactyly, and bilateral short fifth metacarpals. At radiographs, partial or complete fusion of the fourth and fifth metacarpals are diagnosed. The anomaly occurs as an isolated trait or part of a genetic syndrome. Isolated MF4 has been reported in many families, with X-linked recessive inheritance in most of them [1-6], and autosomal dominant mode in one family [7]. Metacarpal fusion of 4-5 was reported associated to metatarsal synostosis and syndactyly, in which missense mutations in HOXD13 gene have been reported in a large Chinese family.
Jamsheer et al (2013) performed an exome sequencing for two unrelated patients originated from Poland and Germany and identified two nonsense mutations in exon 3 of the FGF16 gene: c.C535T; p.R179X and c.C470A; p.S157X [8]. These patients were negative for mutation in the NOG, GDF5, and HOXD13 analyzed before exome sequencing. Recently, other mutations were reported in five other patients from different ethnicity in the three exons of the FGF16 gene. We report here the first Moroccan case of MF4, for whom the already reported mutation (c.C535T; p.R179X) was found.
| Case Report | ▴Top |
Clinical report
The patient was a Moroccan consanguineous 18-month-old boy, the third child of a family of three. The parents are German cousins. The older sisters and parents are healthy. He was born at term, with normal mensurations. He had normal psychomotor development. At clinical exam at 18 months, he had normal mensurations. Exam of hands showed ulnar deviation of both fifth fingers (Fig. 1a). Radiographs showed fusion of the fourth and fifth metacarpals and a bilateral short fifth metacarpal (Fig. 1b). He is programmed for osteotomy of metacarpal fusion. He had minor facial dysmorpy with upslanted palpebral fissures, thin upper lip, and bulbous nose. The patient did not have generalized joint laxity or hyperextensibility. His echocardiography was normal.
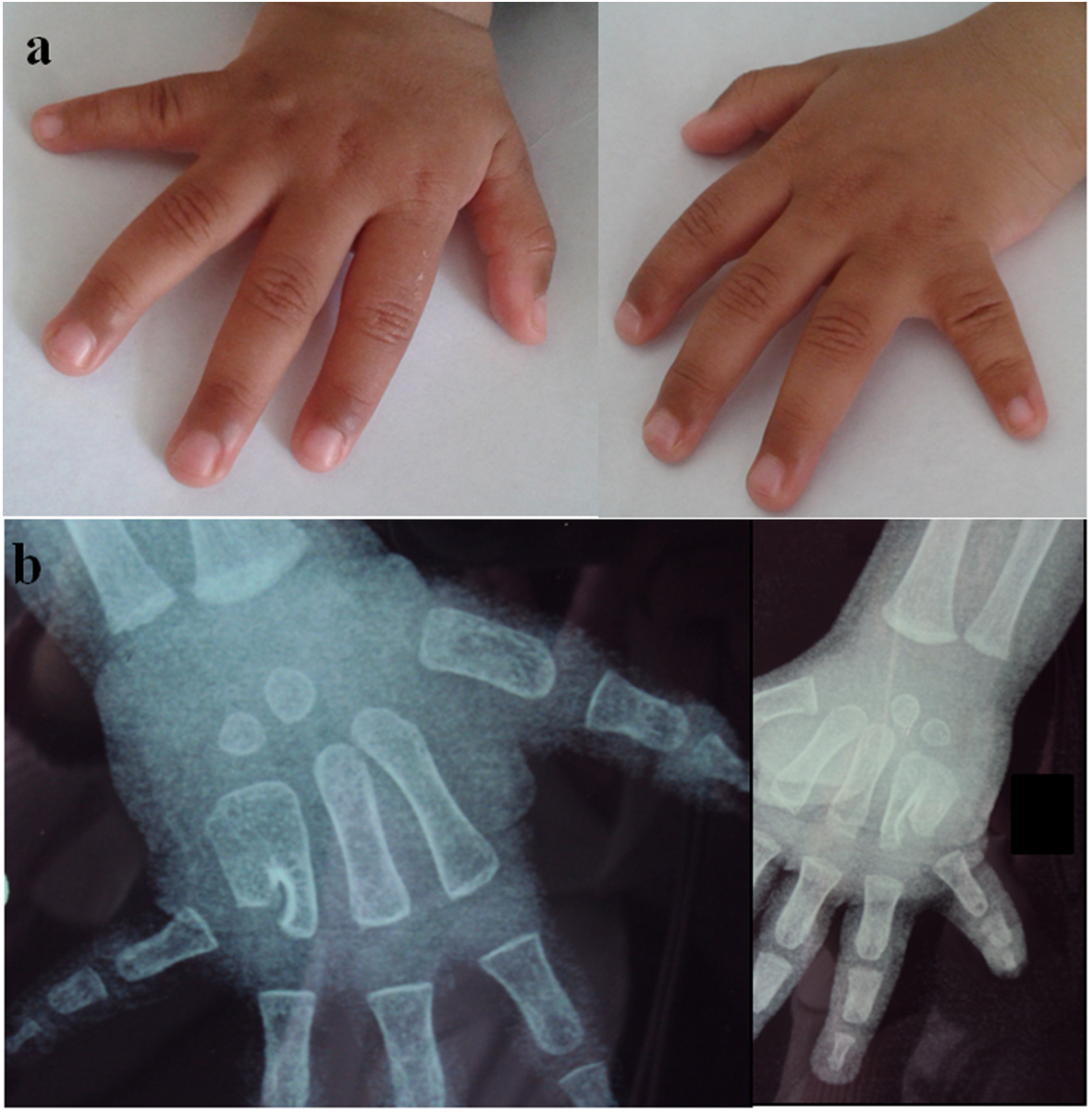 Figure 1. Hand malformation observed in our patient upper limb. (a) Ulnar deviation and shortening of fifth fingers seen upon clinical assessment. (b) Radiograph of the hands showing bilateral partial fusion of metacarpals 4 and 5 and bilateral shortening of fifth metacarpals. Figure 1. Hand malformation observed in our patient upper limb. (a) Ulnar deviation and shortening of fifth fingers seen upon clinical assessment. (b) Radiograph of the hands showing bilateral partial fusion of metacarpals 4 and 5 and bilateral shortening of fifth metacarpals. |
Molecular screening
Informed consent was obtained from the proband’s parents prior to implementation of the genetic studies reported here. Peripheral blood was collected from the affected child and their parents. Genomic DNA was extracted from blood using QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA).
Sanger sequencing of the entire coding sequence of FGF16 gene was performed. We searched for mutations in each of the coding exons and their intronic borders of FGF16 gene. PCR of the three exons of the gene has been realized, and the products were sequencing with BigDye Terminator on an ABI 3130 sequencer (ABI).
Primers for all three exons of human FGF16 gene and their flanking intronic regions were used [8]. Because exon 1 was not annotated in UCSC and Ensemble databases, DNA sequence numbering was based on annotated chromosome X genomic scaffold (NW_004078123.1). Translated protein sequence was identical with the RefSeq entry NP_003859.
The mutation in coding exon 3, c.C535T; p.R179X resulting in codon stop in amino acid 179 was detected in the patient. The mother was found in heteorozygote state (Fig. 2).
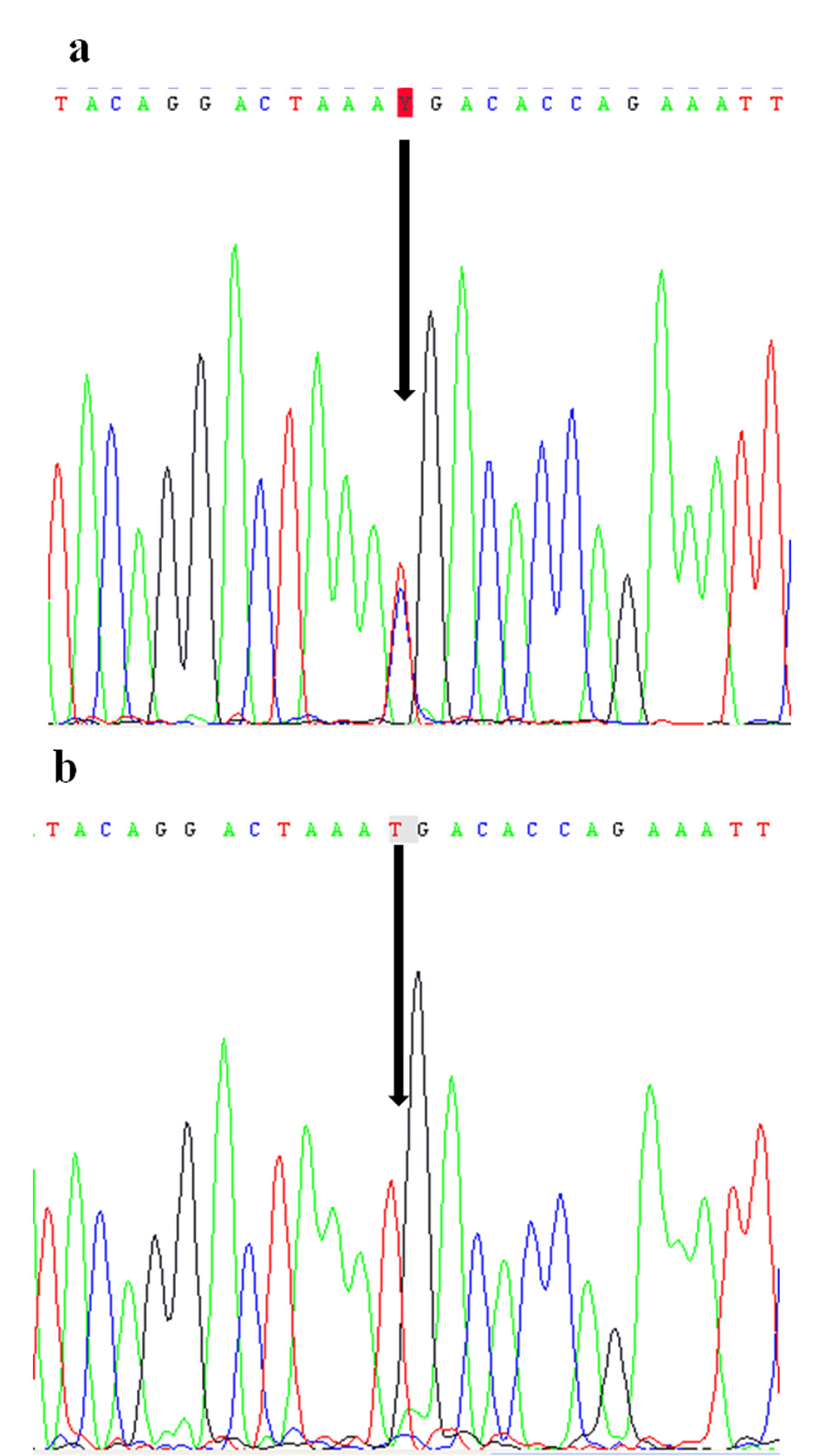 Figure 2. Electropherograms of the nonsense mutation c.C535T (p.R179X) identified in FGF16 gene: in heterozygote state in the patient (a) and in hemizygous state in his mother (b). Figure 2. Electropherograms of the nonsense mutation c.C535T (p.R179X) identified in FGF16 gene: in heterozygote state in the patient (a) and in hemizygous state in his mother (b). |
| Discussion | ▴Top |
Metacarpal 4-5 fusion is a rare congenital malformation of the hand. It was reported to be X-linked recessive. X-linked recessive inheritance was first suggested by Orel (1928) and Holmes et al in 1972 who described first the fusion of the fourth and fifth metacarpals as an entity [3, 6]. Lonardo et al (2004) described a four-generation family with fusion of the fourth and fifth metacarpals, and only males were affected [5].
In this report, we present a further case of sporadic male case of MF4 with recurrent nonsense mutation in the exon 3 of the FGF16 gene. Seven patients with MF4, and mutations in FGF16 gene were reported to date. The first patient was a Polish boy who had a c.535C > T transition in exon 3 of the FGF16 gene, resulting in an arg179-to-ter (R179X) substitution. His clinically and radiologically unaffected mother had the same mutation in heterozygosity state. The second patient originated from Germany had the c.470C > A mutation in the same exon, resulting in a ser157-to-ter (S157X) substitution [8]. The mutation was predicted to produce a protein lacking some of the amino acid residues responsible for receptor and heparan sulfate binding. Jamsheer et al (2014) reported a third patient with generalized joint laxity and hypermobility, associated with MF4 symptoms. This patient had a novel truncating mutation (c.474_477del; p.E158DfsX25) in exon 3 of the gene [9]. He suggested that FGF16 gene may also be responsible for connective tissue symptoms in MF4 patients.
Recently, other mutations were reported in four other patients from different ethnicity. Laurell et al identified three novel mutations [10]. Two of these mutations were reported in unrelated Sweden patients. The first Sweden patient with a familial history of MF4 and cardiac disorders had the c.361G > T (p.G121*) nonsense mutation in exon 2 of FGF16. The second had the c.203G > T (p.R68L) in exon 1. He reported a third patient originated from South America with the c.378G > C (p.S126S) mutation in exon 2 [10]. Eight FGF16 mutation-carrying members of the large first family reported by Laurell et al had reported cardiac symptoms, like myocardial infarction and atrial fibrillation. These findings confirm the variable expressivity of the MF4 phenotype.
Variable expression was reported, ranging from isolated bilateral fusion of metacarpals 4/5 to a broad proximal fifth metacarpal bone and an overall shorter fifth digit. Previously Lu et al reported that that Fgf16 is required in embryonic heart development in mice [11]. Furthermore, Laurell et al reported that 76% of 101 zebrafish embryos studied had a heart edema. Recently, Jones et al reported the seventh case, a fourth British boy with a novel hemizygous 19-bp duplication (c.275_293dup) in exon 2 [12]. Table 1 summarizes the clinical and molecular findings of the previously reported MF4 patients with FGF16 mutations. FGF16 is one of 22 proteins of the fibroblast growth factor family (FGFs) [13]. This gene belongs to the FGF-9 subfamily (composed of FGF9, FGF16, and FGF20) and composed of 621 bp and his protein comprised 207 amino acids [14]. The FGF16 protein has a heparin binding site and a receptor interaction site. The FGFs are a family of growth factors and oncogenes. They play important roles in embryonic development, morphogenesis, tissue repair, inflammation, and tumor growth [15, 16]. FGF16 is expressed predominantly in adult heart tissues [17]. Mouse in situ hybridization confirmed that expression of Fgf16 is also observed in the central interdigital areas of the fore- and hindlimbs [8]. FGFs could be functional by binding and activating FGF receptors (FGFR1 to FGFR4) using their heparin binding site. FGF signaling is regulated by spatial and temporal expression of ligands, heparan sulfate cofactors, receptors, and FGF-FGFR binding specificity [18, 19].
| Table 1. Clinical and Molecular Findings of the Reported FGF16 Mutated Patients With MF4 |
Mutations identified to date, recurrent mutation p.R179X, p.S157X and p.E158DfsX25 mutations introduce premature codons stop in the last exon of the FGF16 gene. Jamsheer et al (2013) proposed that transcripts of these mutations located in the last exon are likely to be stable and not subjected to nonsense mediated decay [8]. The patient reported by Jamsheer with mutation (p.R179X) had joint laxity. Our patient did not have connective tissue disorder symptoms. We can suggest that maybe because the patient is too young these signs are not currently detected. More studies are needed to confirm the impact of impaired FGF16 function in connective tissue properties in human MF4 patients. Our report confirms that mutations of FGF16 are causative for X-linked recessive metacarpal 4-5 fusion.
Acknowledgement
We are grateful to the patient and his family for participating in this study.
| References | ▴Top |
- Anneren G, Amilon A. X-linked recessive fusion of metacarpals IV and V and hypoplastic metacarpal V. Am J Med Genet. 1994;52(2):248-250.
doi pubmed - Habighorst LV, Albers P. [Familial synostosis of metacarpi IV and V]. Z Orthop Ihre Grenzgeb. 1965;100(4):521-525.
pubmed - Holmes LB, Wolf E, Miettinen OS. Metacarpal 4-5 fusion with X-linked recessive inheritance. Am J Hum Genet. 1972;24(5):562-568.
pubmed - Hooper G, Lamb DW. Congenital fusion of the little and ring finger metacarpal bones. Hand. 1983;15(2):207-211.
doi - Lonardo F, Della Monica M, Riccardi G, Riccio I, Riccio V, Scarano G. A family with X-linked recessive fusion of metacarpals IV and V. Am J Med Genet A. 2004;124A(4):407-410.
doi pubmed - Orel H. Kleine Beitrage zur Vererbungswissenschaft. Synostosis metacarpi quarti et quinti. Z Anat. 1928;14:244-252.
- Temtamy SA, McKusick VA. The genetics of hand malformations. Birth Defects Orig Artic Ser. 1978;14(3):i-xviii, 1-619.
pubmed - Jamsheer A, Zemojtel T, Kolanczyk M, Stricker S, Hecht J, Krawitz P, Doelken SC, et al. Whole exome sequencing identifies FGF16 nonsense mutations as the cause of X-linked recessive metacarpal 4/5 fusion. J Med Genet. 2013;50(9):579-584.
doi pubmed - Jamsheer A, Smigiel R, Jakubiak A, Zemojtel T, Socha M, Robinson PN, Mundlos S. Further evidence for FGF16 truncating mutations as the cause of X-linked recessive fusion of metacarpals 4 / 5. Birth Defects Res A Clin Mol Teratol. 2014;100(4):314-318.
doi pubmed - Laurell T, Nilsson D, Hofmeister W, Lindstrand A, Ahituv N, Vandermeer J, Amilon A, et al. Identification of three novel FGF16 mutations in X-linked recessive fusion of the fourth and fifth metacarpals and possible correlation with heart disease. Mol Genet Genomic Med. 2014;2(5):402-411.
doi pubmed - Lu SY, Sheikh F, Sheppard PC, Fresnoza A, Duckworth ML, Detillieux KA, Cattini PA. FGF-16 is required for embryonic heart development. Biochem Biophys Res Commun. 2008;373(2):270-274.
doi pubmed - Jones B, Byers H, Watson JS, Newman WG. Identification of a novel familial FGF16 mutation in metacarpal 4-5 fusion. Clin Dysmorphol. 2014;23(3):95-97.
doi pubmed - Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20(11):563-569.
doi pubmed - Miyamoto M, Naruo K, Seko C, Matsumoto S, Kondo T, Kurokawa T. Molecular cloning of a novel cytokine cDNA encoding the ninth member of the fibroblast growth factor family, which has a unique secretion property. Mol Cell Biol. 1993;13(7):4251-4259.
pubmed - Martin GR. The roles of FGFs in the early development of vertebrate limbs. Genes Dev. 1998;12(11):1571-1586.
doi pubmed - Mariani FV, Ahn CP, Martin GR. Genetic evidence that FGFs have an instructive role in limb proximal-distal patterning. Nature. 2008;453(7193):401-405.
doi pubmed - Miyake A, Konishi M, Martin FH, Hernday NA, Ozaki K, Yamamoto S, Mikami T, et al. Structure and expression of a novel member, FGF-16, on the fibroblast growth factor family. Biochem Biophys Res Commun. 1998;243(1):148-152.
doi pubmed - Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell. 2000;101(4):413-424.
doi - Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252(5013):1705-1708.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.