

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 2, Number 2, December 2013, pages 78-82
Importance of Ultrasound Imaging in Pheochromocytoma in a Child: Case Report and Review of Literature
Nida Dincela, e, Ipek Kaplan Buluta, Kadriye Ozdemira, Mustafa Orhan Bulutb, Betul Sozeria, Z. Hasan Gunc, Sevgi Mira, Hudaver Alperd
aPediatric Nephrology, Ege University Faculty of Medicine, Izmir, Turkey
bPediatric Cardiology, Ege University Faculty of Medicine, Izmir, Turkey
cEge University Faculty of Medicine, Izmir, Turkey
dRadiology, Ege University Faculty of Medicine, Izmir, Turkey
eCorresponding author: Nida Dincel, Pediatric Nephrology, Ege University Faculty of Medicine, Izmir, Turkey
Manuscript accepted for publication October 18, 2013
Short title: Ultrasound Imaging in Pheochromocytoma
doi: https://doi.org/10.4021/ijcp122e
| Abstract | ▴Top |
Pheochromocytomas are rare tumors, originating from adrenal medulla (80-90%), diagnosed at 0.1% of cases with hypertension. Not very rare (40%), diagnosis can be insidious during ultrasound imaging (US) for a non-specific complaint. We are presenting a 13 years old boy who had suffered from non-specific headache without hypertension and any of autonomic disorders, diagnosed by increased urine catecholamine after a mass seen in US. A 13 years old boy has admitted to emergency with blood pressure measurement of 160/110 mmHg. He had severe headache for 18 months without hypertension. In physical exam, hypertension, tachycardia, tremor, and Marfanoid morphology were found. Ultrasound revealed an incidental mass of 33 х 68 mm, with increased vascularization, on pre- and para-aortic sides. Urinary catecholamine and metabolites were grossly elevated. Iodine-131-metaiodobenzylguanidine scintigraphy also showed the heterogeneous mass located in the right para-aortic region, neighboring the liver. Pheochromocytoma is a rare catecholamine-producing tumor, and has life-threatening consequences. Clinical manifestations can be non-specific. It can be easily diagnosed if it is suspected or screened by US. Here we want to emphasize that such an easy and cheap method (screening by US) can lead us to know earlier this high complicated and incidental disease.
Keywords: Pheochromocytoma; Childhood; Hypertension; Ultrasound
| Introduction | ▴Top |
Paragangliomas are rare neuroendocrine tumors, originating from adrenal medulla or extra-adrenal paraganglia. Mostly (80-90%) they are placed in adrenal medulla and thereby called pheochromocytoma. Extra adrenal ones are termed as paragangliomas. Not very rare (40%), diagnosis can be insidious during ultrasound imaging (US) for a non-specific complaint [1, 2]. Pheochromocytomas are diagnosed at approximately 0.1% of cases with arterial hypertension. Twenty-five to sixty percentages of extra adrenal tumors cause headache and paroxysmal hypertension via catecholamine secretion [3]. Their clinical presentation includes episodic headache, diaphoresis, and tachycardia. We are presenting a 13 years old boy suffering from headache for 18 months without hypertension, hospitalized for hypertensive attack, diagnosed with US as pheochromocytoma.
| Case Report | ▴Top |
A 13 years old boy has admitted to the emergency due to nasal bleeding. The measured values of the blood pressure (BP) were 160/110 mmHg. He was directed for hospitalization urgently with signs of hypertensive crisis. In the history, we learned that he had a severe headache for 18 months without hypertension. There was nothing important either in his birth and childhood or family history. In his physical exam, BP of 170/100 mmHg, heart rate of 140/min, non-arrhythmic, tremor in his hands, and asthenic body that gives him a Marfanoid morphology (Fig. 1) were found. Body mass index was within the normal range at 20.5. There were no palpable abdominal mass and no pathologic differences among the four extremities BP. All measures of BP were elevated at 180/100 mmHg with no orthostatic changes. In laboratory evaluation, pathological values were as follows: fasting blood glucose, 142 mg/dL; red blood cell (RBC), 5.2 million/mm3; white blood cell (WBC): 13.2 х 103/mm3; absolute neutrophil count (ANC), 3,400; erythrocyte sedimentation rate (ESR), 65 mm/30 min (Table 1).
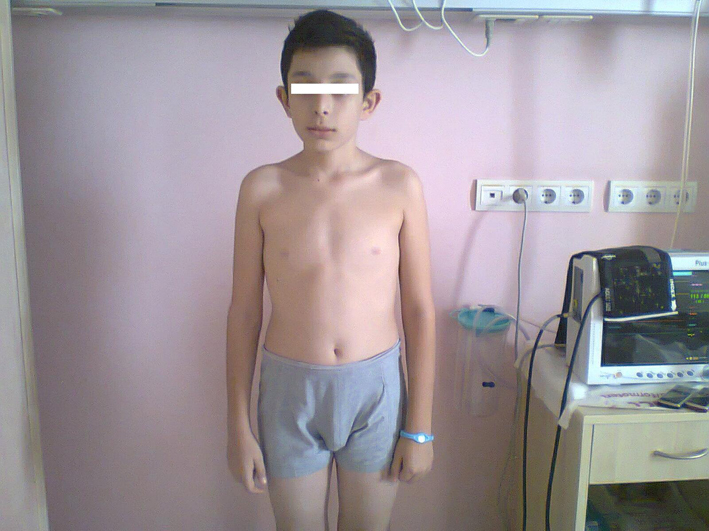 Click for large image | Figure 1. Features of patient. |
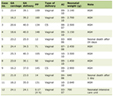 Click to view | Table 1. Preoperative and Postoperative Pathologic Measurements: Laboratory Measurements |
Renal ultrasound examination, done by a digital Compaq 35,000 Hz wavelength US device, revealed an incidental mass of 33 х 68 mm, with an increased vascularization, on the pre- and para-aortic sides. In this way, instead of researching all diagnostic parameters, we awayed to the diagnosis of pheochromocytoma earlier than others. A subsequent magnetic resonance confirmed a solid lobular mass, located in dorsal pancreas, enveloping the renal artery, pushing renal vein up (Fig. 2). No regional adenopathy was evident. There were no other features suggestive of multiple endocrine neoplasia. Urinary collection over 24 h revealed grossly elevated levels of catecholamine and metabolites (metanephrine: 1,278 µg, vanil mandelic acid: 19.3 mg, epinephrine: 747 µg and norepinephrine: 254 µg). Iodine-131-metaiodobenzylguanidine scintigraphy also showed the heterogeneous mass located in the right para-aortic region, neighboring the liver.
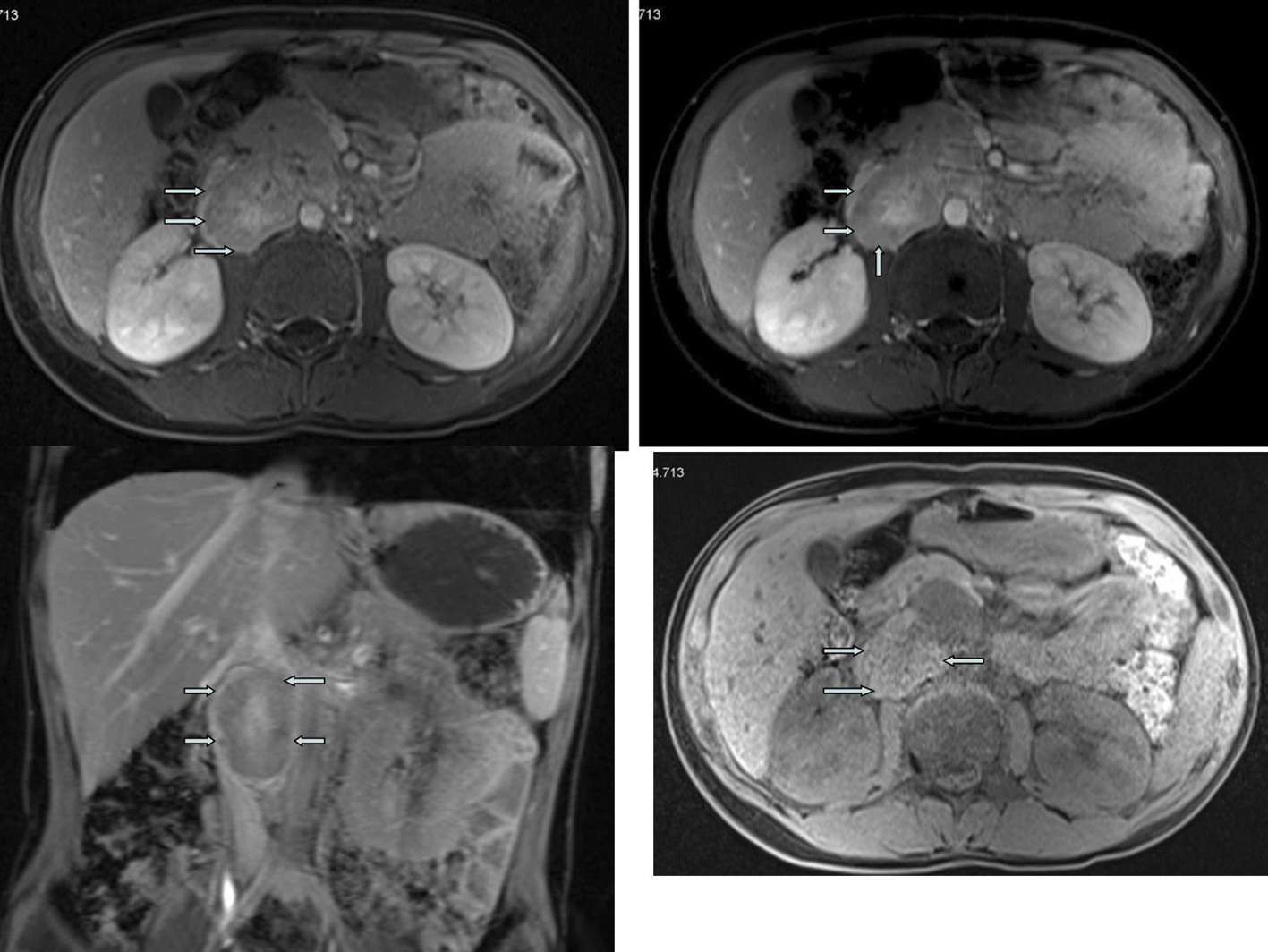 Click for large image | Figure 2. Magnetic resonance imaging. |
Starting after doxazosin (selective α1 blocker) and phenoxybenzamine (α-adrenergic blockage) combination, BP showed slight tendency of lowering, but was stable at levels of 130 - 140 mmHg for systolic blood pressure (SBP) and 90 - 100 mmHg for diastolic blood pressure (DBP). With the stable BP values, he went to operation. Operation material was sent for pathologic exam. Following the operation, our patient’s antihypertensive requirements were significantly reduced. End organ damage was searched. Ventricular mass index and non-nephrotic level of proteinuria were increased. Two weeks postoperatively, ultrasound revealed any mass for residual tumor and metastatic disease. Urinary catecholamine and metabolite concentrations were within the normal range after a follow-up of 3 months. Histopathological examination of the excised tissue confirmed a 5 х 5.5 cm solid heterogeneous tumor invasing renal capsule (Fig. 3). Microscopic evaluation revealed pheochromocytoma, with infiltration of the capsule. Iodine-131-metaiodobenzylguanidine scintigraphy, performed 3 months after operation, confirmed complete excision with no evidence of metastatic disease. Our patient is symptom-free with normal urinary catecholamine and metabolites and normal BP for 3 years (Table 1).
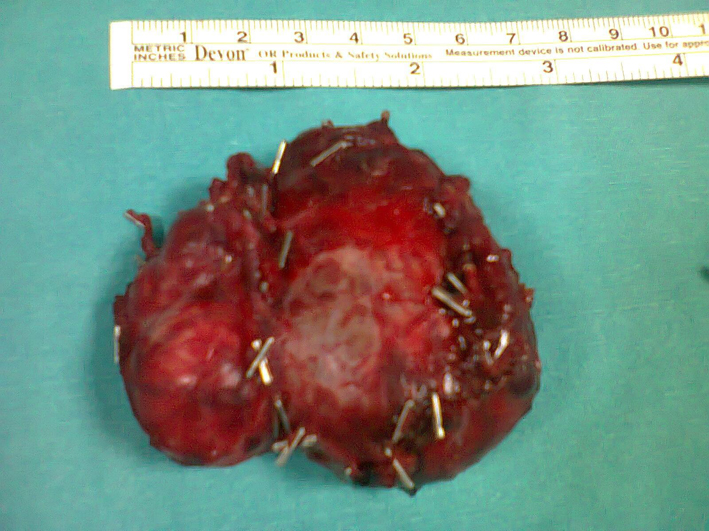 Click for large image | Figure 3. Histopathological evaluation. |
| Discussion | ▴Top |
Pheochromocytoma is a rare, insidious tumor representing approximately 5% of adrenal incidentalomas [4]. It originates from sympathetic paraganglioma of chromaffin cells [5, 6]. Abdominally located paragangliomas are mostly in the retroperitoneal region, on the anterolateral side of abdominal aorta, and on the bifurcation of mesenteric artery with aorta. Clinical presentation shows signs of catecholamine excess: hypertension, tachycardia, headache, sweating, nausea, vomiting, trembling, weakness, irritation, abdominal and chest pain, dyspnea, red warm face, constipation, polyuria, and polydipsia [7].
Among the whole, 20-30% of adrenal pheochromocytomas are asymptomatic; they are called clinically silent pheochromocytoma [8, 9]. These forms are generally larger tumor size at the time of diagnosis, explained by: as the tumor gets larger, symptoms get fewer. This was explained by extensive necrosis area at the tumor center, which may significantly decrease the number of cells producing catecholamine [10]. Similarly in our case, symptoms were not apparent; he had been only suffering from non-specific headache without hypertension and autonomic dysfunctions for 18 months.
The tumor size varies; the largest one was 29 х 21 х 12cm reported in literature [11]. The mean size in operated cases was known as 3 - 5 cm, and generally in malignant cases greater than 6 cm. However, in our case, size of the mass was 33 х 68 cm. It is also known that, a giant tumor with capsular and/or vascular invasion could be benign. Apart from size, postoperative persistence of arterial hypertension was more prone to show malignant character of tumor [12]. In our case, blood pressure was normal postoperatively.
As supported with literature knowledge, besides hypertensive attacks, headache, pallor, palpitation, and sweating are the classical symptoms. Our case had headache attacks without hypertension for 18 months. Also, there were no any of these autonomic dysfunctions such as sweating, trembling, and tachycardia. Therefore, pheochromocytoma was not present in our minds to role out at beginning. For differential diagnosis of hypertension, ultrasound of kidneys was performed and mass was seen. After detecting of adrenal mass by US, the algorithm must be followed, which was given in Figure 4 [13]. As the gold standard diagnostic criteria are catecholamine and their metabolites in urine, urine metabolites of our patient were studied and found significantly high. High serum fasting glucose, sedimentation rate, leukocytosis, and erythrocytosis were also all supported diagnosis as well as in the literature data [14].
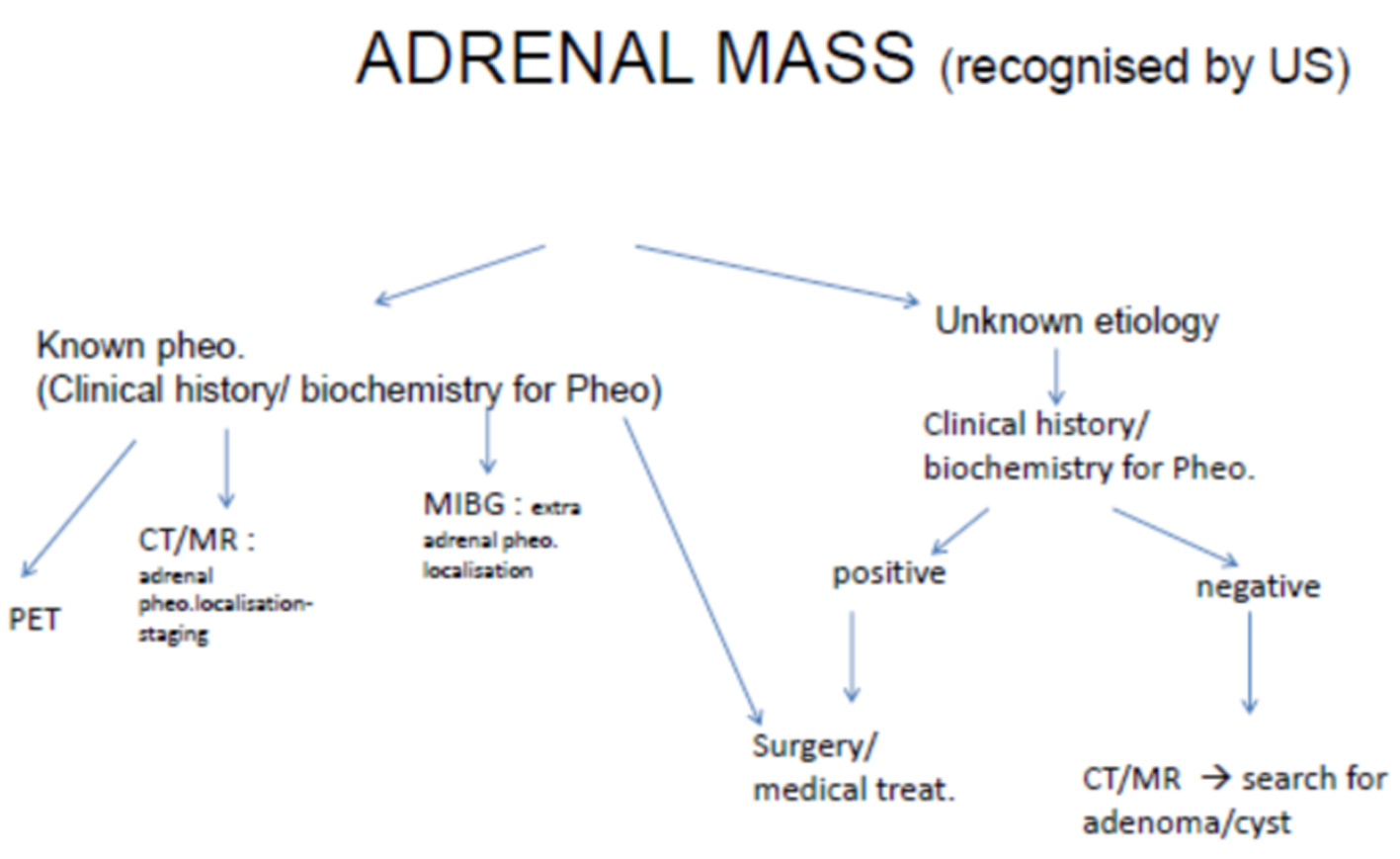 Click for large image | Figure 4. Diagram illustrates algorithm for the assessment of an adrenal mass and recognition of a pheochromocytoma. |
Surgical resection is the definitive treatment for patients with pheochromocytoma [3]. In our case, an open approach was undertaken; during operation hypertensive attacks and hypotensive reactions were seen because of catecholamine secretion. It is recommended that all patients receive 7 to 10 days of α-adenoreceptor antagonists (phenoxybenzamine) preoperatively to prevent catecholamine induced vasoconstriction and β blocker must be added if tachycardia occurs. Due to the risk of vasoconstriction-aggravated hypertension, β blockers must not give before having α-blockadge. Also in order to prevent hypertension attacks, α-adenoreceptor antagonists, calcium-channel blockers (especially in labile hypertension) or angiotension-receptor blockers can be used preoperatively [15]. During operation nitroprusside infusion may be needed. We had controlled hypertension of our case with phenoxybenzamine and doxazosin, no need for β blocker preoperatively and during operation given with nitroprusside infusion.
It is important to be aware of the mass earlier than the research for hypertension etiologies. In a study of a case reported by Yuksel et al, they notified the importance of hypertension etiology and easily search with US [16].
Major consequences of the disease are besides malignant potential, permanent hypertension with end organ damage, hyperglycemia, glycosuria, and sudden hypotension. Our case had end organ damages secondary to chronic hypertension.
Conclusion
Pheochromocytoma is a rare catecholamine-producing tumor with potential life-threatening consequences. Clinical manifestations can be atypical and non-specific. It can be easily diagnosed if it is suspected or screened by US. In our case US revealed the diagnosis, although for the certainty of diagnosis, we applied other more specified and sensitized imaging tools. Here we want to emphasize that such an easy and cheap method (screening by US) can lead us to know earlier this high complicated and incidental disease.
1) Rare but life-threatening disease
2) Atypical clinical manifestations
3) Can diagnosed earlier by US
4) US: clear and cheap method
| References | ▴Top |
- Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23(34):8812-8818.
doi pubmed - Reisch N, Peczkowska M, Januszewicz A, Neumann HP. Pheochromocytoma: presentation, diagnosis and treatment. J Hypertens. 2006;24(12):2331-2339.
doi pubmed - Dluhy RG and Gittes. Feokromositoma: the adrenals, in Campbell’s Urology, Eds. by: P.C.Walsh.RF, Gittes, AD, Perimutter, TA, Starney, fifthed. vol:3, chap:85.W.B.Saunders Co. London. pp:3027.1986.
- Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Ali A, Giovagnetti M, et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000;85(2):637-644.
doi pubmed - Tischler AS, Kimura N, McNicol AM. Pathology of pheochromocytoma and extra-adrenal paraganglioma. Ann N Y Acad Sci. 2006;1073:557-570.
doi pubmed - Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675.
doi - Marinov L. Phechromocytoma during childhood - a case report.Journal of IMAB -Annual Proceeding (Scientific Papers). 2009; book1.
- Andreoni C, Krebs RK, Bruna PC, Goldman SM, Kater CE, Alves MT, Ortiz V. Cystic phaeochromocytoma is a distinctive subgroup with special clinical, imaging and histological features that might mislead the diagnosis. BJU Int. 2008;101(3):345-350.
doi pubmed - Antedomenico E, Wascher RA. A case of mistaken identity: giant cystic pheochromocytoma. Curr Surg. 2005;62(2):193-198.
doi pubmed - Li C, Chen Y, Wang W, Teng L. A case of clinically silent giant right pheochromocytoma and review of literature. Can Urol Assoc J. 2012;6(6):E267-269.
pubmed - Basso L, Lepre L, Melillo M, Fora F, Mingazzini PL, Tocchi A. Giant phaeochromocytoma: case report. Ir J Med Sci. 1996;165(1):57-59.
doi pubmed - Goldstein DS, Eisenhofer G, Flynn JA, Wand G, Pacak K. Diagnosis and localization of pheochromocytoma. Hypertension. 2004;43(5):907-910.
doi pubmed - Blake MA, Kalra MK, Maher MM, Sahani DV, Sweeney AT, Mueller PR, Hahn PF, et al. Pheochromocytoma: an imaging chameleon. Radiographics. 2004;24(Suppl 1):S87-99.
doi pubmed - Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212-215.
doi - Proye C, Thevenin D, Cecat P, Petillot P, Carnaille B, Verin P, Sautier M, et al. Exclusive use of calcium channel blockers in preoperative and intraoperative control of pheochromocytomas: hemodynamics and free catecholamine assays in ten consecutive patients. Surgery. 1989;106(6):1149-1154.
pubmed - Yuksel H, Odabasi AR, Onur E. Gebelikte feokromasitoma: olgu sunumu. ADU Tip Fakultesi Dergisi. 2002;3(2):29-31.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.